
 No English version available for this Article No English version available for this Article |
| Rev Esp Endocrinol Pediatr 2011;2 Suppl(1):75-78 | Doi. 10.3266/RevEspEndocrinologPediatr.pre2011.May.66 |
| Tiroides |
| Sent for review: 2 May. 2011 | Accepted: 3 May. 2011 | Published: 3 May. 2011 |
| Congreso SEEP |
| Correspondence: Congreso SEEP E-mail: seep@seep |
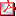 Tabla - O1d2-005_013_Hipotiroidismo Congénito y TNF a 0, 3, 6 meses Tabla - O1d2-005_013_Hipotiroidismo Congénito y TNF a 0, 3, 6 meses |
|
|
O1/d2-005 HIPOTIROIDISMO CONGÉNITO PRIMARIO (HC): NORMALIZACIÓN DE LOS VALORES DE TNF-α CON EL TRATAMIENTO CON LEVOTIROXINA. B. Huidobro Fernández (1), M. Echeverría Fernández (1),
(1) Unidad de Metabolismo/Endocrinología Pediátrica. Hospital General Universitario Gregorio Marañón. Madrid, (2) Laboratorio de Inmunobiología Molecular. Hospital General Universitario Gregorio Marañón. Madrid.
Introducción: El TNF-α participa en procesos de activación, proliferación, diferenciación y apoptosis glial y neuronal, desempeñando un papel fundamental tanto en el desarrollo del sistema nervioso central como en procesos patológicos. Las hormonas tiroideas son necesarias para el desarrollo cerebral normal. Previamente se ha comunicado que los niveles de TNF-α son indetectables en recién nacidos con HC.
Objetivo: Determinar los niveles de TNF-α en pacientes con HC antes de iniciar tratamiento y tras la normalización de la función tiroidea.
Sujetos y Métodos: Determinación de TNF-α, TSH y T4L en pacientes diagnosticados de HC antes de iniciar tratamiento con levotiroxina (basal), a los 3 y a los 6 meses. Determinación TSH/T4L: inmunoquimioluminiscencia. Cuantificación TNF-α: inmunoensayo. Análisis estadístico: SPSS 15.0, t de Student.
Resultados: Se incluyeron 21 pacientes (9 niñas, 12 niños). Edad al diagnóstico: mediana 9 días (rango 6-25). Gammagrafía tiroidea Tc99: ausencia de captación (4), ectopia sublingual (6), eutópicos (11). Los resultados se presentan en la Tabla 1. En 19 pacientes (90%) los niveles de TNF-α fueron <4 pg/ml en la primera determinación y todos ellos presentaron elevación del TNF-α en la segunda determinación (hubo 2 pacientes en los que no se elevó, fueron los 2 que tuvieron determinación inicial normal), siendo la diferencia estadísticamente significativa. En la tercera determinación todos los pacientes presentaron TNF-α ≥4 pg/ml excepto 2 pacientes (3.67 y 3 pg/ml).
Conclusiones: Los niveles de TNF-α se encuentran disminuidos al diagnóstico en los pacientes con HC y se normalizan con el tratamiento con levotiroxina. El TNF-α podría ser uno de los mecanismos implicados en las alteraciones del SNC asociadas al HC y su monitorización puede ser útil en la optimización terapéutica de los pacientes con HC
O1/d2-006 EL ARRAY-CGH “DIRIGIDO” THYROARRAY-V1.0 REVELA DEFECTOS GENÉTICOS EN EL HIPOTIROIDISMO CONGÉNITO NO IDENTIFICABLES POR PCR Y SECUENCIACIÓN. C.M. Moya (1), E. Vallespín (2), M. Polak (3), D. Kariyawasam (3), P. Lapunzina (2), J. Nevado (2), J.C. Moreno (1).
(1) Laboratorio Molecular de Tiroides. Instituto de Genética Médica y Molecular. Hospital Universitario La Paz. Madrid, (2) Laboratorio de Genómica Estructural. INGEMM. Hospital Universitario La Paz. Madrid, (3) Endocrinología Pediátrica. Hôpital Necker-Enfants Malades, París, Francia.
En la actualidad se conocen defectos en 26 genes que conducen a Hipotiroidismo Congénito (HC). Sin embargo, sigue siendo reducido el porcentaje de pacientes con mutaciones patogénicas que explican definitivamente el fenotipo clínico. Esto puede indicar la existencia de otros genes responsables de HC aún sin identificar. Sin embargo, el screening mutacional se realiza mayoritariamente con técnicas de PCR y secuenciación Saenger, que presentan claras limitaciones.
Objetivo: Testar la capacidad de los arrays de Hibridación Genómica Comparada (CGH) para identificar deleciones o duplicaciones en pacientes con hipotiroidismo molecularmente no filiado tras screening mutacional clásico.
Métodos: Diseño personalizado de un array-CGH dirigido
Resultados: Se identificaron deleciones heterocigotas en el gen de tiroglobulina de 1.2 y 10 Kbp que eliminan respectivamente los exones 20 y 45 en 2 pacientes. Una deleción de 50 bp que elimina parte del exón 16 de DUOX2 en un paciente con una única mutación A1277G identificada en el mismo gen. Una duplicación introexónica que disrupciona el exón 5 de DUOX2 y una deleción de 470 bp que elimina la región codificante del exón 12 de PAX8. La predicción patogénica de estas aberraciones genéticas es muy alta.
Conclusiones: El array-CGH de diseño “personalizado” de tiroides detecta defectos genéticos que se escapan al estudio clásico de mutaciones. La gran densidad de oligos permite la identificación de deleciones/duplicaciones muy pequeñas que incluso arrays genómicos comerciales (Agilent 44K y 244K) no hubiesen detectado. Las nuevas técnicas genómicas se hacen indispensables en caso de detección fallida de alteraciones genéticas en los genes clásicos de la hormonogénesis tiroidea. O1/d2-007 ENFERMEDAD DE HIRSCHSPRUNG Y ALTERACIONES EN EL PROTOONCOGEN RET. M.A. Molina Rodríguez (1), E. Bermúdez de Castro López (1), A. De La Puente Arévalo (1), G. Prieto Bozano (2), I. González Casado (1), R. Gracia Bouthelier (1).
(1) Servicio de Endocrinología Infantil, Hospital Universitario La Paz, Madrid; (2) Servicio de Gastroenterología Infantil, Hospital Universitario La Paz, Madrid.
Introducción: Las neoplasias endocrinas múltiples tipo 2 (MEN2), de herencia autosómica dominante, van asociadas a alteraciones en el protooncogen RET. El MEN2 lo podemos subdividir en: MEN2A (carcinoma medular de tiroides, feocromocitoma, hiperplasia o adenoma paratiroideo y, con frecuencia, enfermedad de Hirschsprung), MEN2B (carcinoma medular de tiroides, feocromocitoma, neuromas cutáneos y hábito marfanoide) y carcinoma medular de tiroides familiar (CMTF) (carcinoma medular de tiroides y en ocasiones Hirschsprung).
Objetivos: Revisión de los pacientes diagnosticados de enfermedad de Hirschsprung en los que posteriormente se ha confirmado la presencia de mutación en el gen RET. Se han recogido datos sobre los antecedentes familiares, patologías asociadas, resultado del estudio genético, seguimiento y tratamiento realizado (tiroidectomía y anatomía patológica).
Resultados: Se han estudiado cinco pacientes con enfermedad de Hirschsprung y mutación confirmada en el gen RET (en dos de ellos asociada al MEN2A, en otro al CMTF y los dos últimos presentan una mutación de novo). Cuatro de los pacientes tenían una afectación intestinal amplia (colon e ileon terminal) y en los cinco el despistaje de carcinoma medular de tiroides, hiperparatiroidismo y feocromocitoma fue negativo. Se ha realizado tiroidectomía profiláctica en tres de los pacientes, a las edades de 4,8, 5 y 13 años, y en los otros dos está pendiente.
Conclusiones: El 82% de los pacientes con enfermedad de Hirschsprung con afectación total colónica presentan alteración en el protooncogen RET y el 33% si el segmento afecto es corto. Por tanto, ante el diagnóstico de enfermedad de Hirschsprung, se debe considerar la realización de un estudio genético para descartar un síndrome MEN2. Por otra parte, la prevalencia del cáncer medular tiroideo en pacientes con MEN2 es mayor del 90%, por lo que, ante la confirmación genética de la enfermedad, es obligado realizar una tiroidectomía profiláctica. O1/d2-008 DETECCIÓN DE YODOTIROSINAS EN MUESTRAS BIOLÓGICAS HUMANAS: ¿CROMATOGRAFÍA LÍQUIDA (HPLC) AISLADA O EN TÁNDEM CON ESPECTROMETRÍA DE MASAS (LC/MS)? M. Sanz Rodríguez (1), A. Hernanz Macías (2), A. Buño Soto (2), JC Moreno Navarro (1).
(1) Laboratorio Molecular de Tiroides. INGEMM-Instituto de Genética Médica y Molecular. Hospital Universitario La Paz. Madrid; (2) Servicio de Bioquímica y Análisis Clínicos. Hospital Universitario La Paz. Madrid.
El enzima tiroideo DEHAL1 recicla yodo a través de la desyodación de mono- y diyodotirosinas (MIT, DIT) para la síntesis mantenida de hormonas tiroideas. Los defectos genéticos en DEHAL1 producen un tipo de hipotiroidismo congénito que puede no ser detectado en el screening neonatal, con el consiguiente riesgo de retraso mental en los niños. El diagnóstico específico de este HC es la elevación de MIT y DIT en plasma y orina. En centros de investigación especializados se ha utilizado un radioinmunoensayo (RIA) para determinar DIT. En la actualidad no existe método rutinario de determinación de yodotirosinas aplicable al diagnóstico, ni tampoco al screening metabólico neonatal en papel de filtro.
Objetivo: Determinar la viabilidad de una determinación precisa de ambas yodotirosinas por cromatografía líquida de alto rendimiento (HPLC) o bien la necesidad de la espectrometría de masas para discriminar niveles normales y patológicos de MIT y DIT.
Métodos: Se utilizó un cromatógrafo HPLC Waters 2695 y un detector UV (Waters 2487 Dual Absorbance detector) y una columna C18 para fase reversa (Aminoacid physiologic Pico-Tag®). El análisis se realizó con el software Empower de Waters.
Resultados: El método Pico-Tag® detecta picos de MIT y DIT en solución acuosa con tiempos de retención de 61 y 52 min en rangos de concentración mili-Molar. Para testar la sensibilidad y la capacidad de discriminación del método en muestras bioquímicamente más complejas, se aplicó HPLC a plasmas control dopados con concentraciones conocidas de MIT y DIT en rango nano-Molar, en una curva de calibración inicial de 3 puntos (0, 1 y 10 nM), dentro de los rangos de normalidad descritos en población general para MIT (1-10 nM) y DIT (1-4 nM). En estos rangos de concentración, los picos de MIT y DIT se encuentran solapados por los aminoácidos lisina y fenilalanina, cuyas concentraciones son 1.000 veces superiores a las de las moléculas investigadas. Conclusiones: El método de HPLC comercial utilizado no es capaz de obtener una sensibilidad/especificidad adecuadas para detectar MIT y DIT. El diagnóstico de hiper-yodotirosinemia requiere un mayor rendimiento analítico, como el que actualmente ofrecen las nuevas técnicas de espectrometría de masas en tándem. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites