| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):163-175 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.186 |
| Crecimiento |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
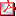 Tabla - P1/d3-022 Tabla - P1/d3-022 |
| Tabla - P1/d3-023 |
| Tabla - P1/d3-024 |
| Tabla - P1/d3-025 |
| Tabla - P1/d3-026 |
| Tabla - P1/d3-027 |
| Tabla - P1/d3-030 |
| Tabla - P1/d3-031 |
| Tabla - P1/d3-032 |
| Tabla - P1/d3-038 |
| Tabla - P1/d3-040 |
|
|
P1/d3-021
LA TALLA FAMILIAR CLAVE EN EL ESTUDIO DE CRECIMIENTO A.Aguayo Calcena(1), A. Vela Desojo(2), A. Salomón Estebanez(2), G. Grau Bolado(2), A. Rodríguez Estévez(2), I. Rica Etxebarria(2). (1) Hospital Universitario Cruces, Sección de Endocrinología Pediátrica, CIBERDEM, Barakaldo/ Bizkaia; (2) Hospital Universitario Cruces, Sección Endocrinología Pediátrica. Barakaldo La talla diana (TD) es fundamental para la valoración del crecimiento. Muchos pacientes que consultan por “talla baja” tienen antecedente de talla baja familiar (TBF), siendo menos frecuente este motivo de consulta en familias con tallas por encima de la media. Se presentan 5 pacientes con déficit orgánico de GH, que mostraban talla baja, retraso de la edad ósea (EO) y una velocidad de crecimiento (VC) adecuada antes de establecer el diagnóstico. Tras descartar las patologías no endocrinas, se analizó la GH que resultó insuficiente. La R.N.M. fue patológica en todos. Dado que uno de los requisitos necesarios para solicitar tratamiento con GH es la VC menor a p25, estos pacientes no se habrían beneficiado en este momento del tratamiento. PACIENTE 1: ♂ 9,2 años; Talla:-2,62 SDS; TD:+1,56 SDS; GH tras clonidina e insulina respectivamente: 0.9 ng/ml y 0.9 ng/ml; R.M.N.: Hipoplasia hipofisaria con sección tallo y neurohipófisis ectópica. PACIENTE 2: ♂ 10.5 años; Talla:-2.06 SDS; TD: +2,13 SDS; GH tras clonidina: 3.3 ng/ml; GH tras insulina: 2.2 ng/ml; RMN: Hipoplasia hipofisaria con sección tallo y neurohipófisis ectópica. PACIENTE 3: ♀ 6.6 años; Talla:-3.6 SDS; TD: +1.09 SDS; GH tras clonidina: 6.8 ng/ml; GH tras insulina: 2.6 ng/ml; RMN: Hipoplasia hipofisaria con sección tallo y neurohipófisis ectópica. PACIENTE 4: ♂ 9.1 años; Talla: -2.9 SDS; TD: -0.3 SDS; GH tras clonidina: 2.5 ng/ml; GH tras insulina: 1.5 ng/ml; RMN: Hipoplasia hipofisaria. PACIENTE 5: ♂ 10.5 años; Talla: -2.02 SDS; TD: -0.3 SDS; GH tras clonidina: 6.8 ng/ml; GH tras insulina: 6.7 ng/ml; RMN: Hipoplasia hipofisaria. La VC previa al tratamiento se mantenía en torno al p 25. En los primeros años con GH la VC > p97 en todos [VC en SDS: media +7,5 SDS (r: 4,2-12,36)]. Se mantiene el retraso en la edad ósea. Comentarios: - Destacar la importancia de la TD ya que una talla alta familiar puede llevarnos a infradiagnosticar una deficiencia de GH. - En todos los pacientes se diagnostica el déficit de GH y se encuentra alteración hipofisaria en la RMN. - La respuesta al tratamiento con GH es espectacular en todos los pacientes.
P1/d3-022
DESCRIPCIÓN DE UNA NUEVA MUTACIÓN EN HOMOCIGOSIS DE IGFALS CON RESPUESTA PARCIAL AL TRATAMIENTO CON HORMONA DE CRECIMIENTO RECOMBINANTE A.M. Velásquez Rodríguez(1), M. Ramón Krauel(1), A. Gómez Núñez(2), A. Campos Barros(3). (1) Hospital Sant Joan de Déu, Sección de Endocrinología Pediátrica, Barcelona; Hospital Sant Joan de Déu, Sección de Endocrinología Pediátrica, Barcelona; (2) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz, Madrid; (3) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz, Madrid y Centro de Investigación Biomédica en Enfermedades Raras (CIBERER, U753), Instituto Carlos III, Madrid Introducción: Mutaciones en homozigosis o heterozigosis combinada del gen codificante de ALS (IGFALS;16p13.3) causan déficit del crecimiento postnatal asociado a niveles de IGF-I e IGFBP-3 muy disminuidos, indicando que ALS desempeña una función relevante en la regulación de la biodisponibilidad de IGF-I circulante durante el crecimiento. Caso clínico: Niña en seguimiento desde los 3 años por retraso ponderoestatural. Nacida de padres consanguíneos a las 39 semanas con PN 2900 gr (-1,2DE), LN 48 cm (-1,4DE) y PC 34 cm (-0,34DE). TM 150 cm (-2,34DE) y menarquia a los 11 años; TP 175 cm (-0,36DE) y pubertad normal. TMP 156±5 cm (-1,34±0,83DE). Hipocrecimiento mantenido durante su seguimiento (ver tabla). Estudios iniciales revelaron niveles de IGF-I indetectables y pruebas de estimulación de GH con picos máximos de 2,96 ng/ml (clonidina), 27,5 ng/ml (L-Dopa/carbidopa) y 7,7 ng/ml (glucagón). Tras el inicio de rhGH (5 años 2 meses) se observa mejoría en la talla y VC, con niveles de IGF-1 bajos aunque detectables y niveles de IGFBP-3 indetectables (ver tabla). Tras 26 meses se suspende el tratamiento con rhGH por decisión de la familia lo que provoca una drástica disminución de la VC (ver tabla). A los 10 años, 1 mes inicia pubertad con progresión adecuada. A los 11 años y 2 meses se realizó TTOG por riesgo descrito de insulinoresistencia, evidenciándose intolerancia a HC y glucemias superiores a 200 en puntos intermedios con HbA1C 5,2%. Estudios moleculares: Análisis mutacional de las secuencias codificantes, transiciones intrón/exón y secuencias reguladoras de IGFALS mediante HRM y secuenciación de las variantes detectadas. Resultados: El estudio molecular detectó la mutación puntual, c.682A>G, p.Asn228Asp, no descrita previamente, en el exón 2 de IGFALS, en homozigosis en el índice y en heterozigosis en ambos progenitores. La mutación afecta a un residuo conservado, localizado en el dominio de la región rica en Leu LxxLxLxxN/ CxL y ha sido clasificada como patogénica por las herramientas bioinformáticas PolyPhen 2.2, SIFT y MutationTaster. Conclusiones: Describimos el primer caso femenino en España con hipocrecimiento por una nueva mutación en homozigosis de IGFALS, asociado a deficiencia severa de IGF-I y con respuesta parcial al tratamiento con rhGH.
P1/d3-023
TRATAMIENTO CON HORMONA DE CRECIMIENTO (GH) EN PACIENTES CON SÍNDROME DE SILVER RUSSELL. RELACIÓN GENOTIPO-FENOTIPO Y RESPUESTA AL TRATAMIENTO HASTA INICIO DE PUBERTAD A. Rodríguez Estévez(1), E. Artola Aizalde(2), I. Diez López(3), C. Fernández Ramos(4), N. Portillo Nájera(5), A. Vela Desojo(6). (1) Hospital Universitario de Cruces, Barakaldo; (2) Hospital Universitario Donostia. Donosti; (3) Hospital Universitario de Araba. Vitoria-Gasteiz; (4) Hospital Universitario de Basurto. Bilbao; (5) Hospital Universitario de Cruces. Barakaldo; (6) Hospital Universitario de Cruces. Barakaldo. CIBERER Introducción: El síndrome de Silver-Russell (SR) se caracteriza por CIR, pobre crecimiento postnatal y fenotipo peculiar. El diagnóstico clínico es difícil. La hipometilación en ICR1 de 11p15 es la causa más frecuente (60%). La disomía uniparental materna del cromosoma 7 (mUDP7) está presente en 5-10%. Objetivo: Valorar la respuesta al tratamiento con GH en los 2 primeros años de tratamiento y la modificación en EO por método Greulich-Pyle (GP) respecto a la talla (DE) durante el mismo periodo y al inicio pubertad. Relación del fenotipo con mutación, y del genotipo con los parámetros de crecimiento. Material y métodos: 13 pacientes (6V, 7M) con fenotipo SR seguidos en Comité GH. 4 ICR1 hipometilación (2V, 2M), 3 mUDP7 (2V, 1M) y 1M Duplicación 11p15 de origen materno. Estadística SPSS18 (descriptivo: medias y DE; prueba T para muestras relacionadas). Resultados: Edad de inicio del tratamiento 6.4±2.6 años. Alcanzan pubertad 9 (5V, 4M) a edad media 11.6 y 10.4 años, respectivamente. El hallazgo fenotípico más frecuente fue la macrocefalia relativa/frente abombada y facies triangular. Pacientes con ICR1 hipometilación (2V, 2M) y Duplicación 11p15 materna (1M) asociaban más características del fenotipo clásico que los afectos de mUDP7 (2V, 1M): hemihipertrofia 4/5 vs 0/3; y clinodactilia 4/5 versus 1/3. Retraso en lenguaje/THDA en 2/5 y 1/3 (ICR1 vs mUDP7). Disquinesia mioclónica en 1V mUDP7. AF y AP y Evolución del crecimiento con GH en Tabla 1. Encontramos diferencias significativas entre VC0 y VC1 (p ,001) IC -3.26/-1.15 (95%) y entre VC0 y VC2 (p ,010) IC -3.03/-.52 (95%). No asociación entre genotipo y diferentes variables auxológicas. Conclusiones: 1. Fenotipo clásico más frecuente en ICR1 hipometilación, especialmente la asimetría. Retraso del lenguaje similar en ambos grupos. 2. Mejoría evidente de VC con GH durante los primeros años y hasta la pubertad, a expensas de un avance en la EO.
P1/d3-024
DIAGNÓSTICO DE TALLA BAJA EN UNA CONSULTA DE ENDOCRINOLOGÍA PEDIÁTRICA DE UN HOSPITAL PROVINCIAL P. Sevilla Ramos, M. Alija Merillas, N. López Andrés, G. Arriola Pereda H. Universitario De Guadalajara, Guadalajara Introducción: La talla baja es un diagnóstico frecuente en consulta de endocrinología pediátrica. El conocimiento de las características de la patología atendida es clave para que los distribuidores de salud realicen una adecuada distribución de recursos. Objetivos: Conocer las características de los pacientes diagnosticados de talla baja. Material y métodos: Estudio descriptivo retrospectivo de los datos obtenidos de historias clínicas de pacientes menores de 15 años diagnosticados de talla baja en consulta de endocrinología pediátrica de nuestro hospital provincial entre 1-1-2009 y 31-12-2011, centro de referencia para una población de unos 42000 pacientes menores de 15 años. Variables recogidas: sexo, fecha de nacimiento, edad de primera consulta, diagnóstico siguiendo la clasificación publicada por la ESPE en 2007, número de visitas realizadas en el periodo de seguimiento y tratamiento administrado. Resultados: Durante el periodo de estudio fueron valorados en consulta de endocrinología 989 pacientes, de los cuales 299 fueron diagnosticados de talla baja (30.2%). El 55.2% de los pacientes con talla baja eran varones y el 44.8% mujeres. La edad media fue de 6.94 años (+/-3.9), mediana de 7, no apreciándose diferencia de edad por sexo. Los diagnósticos más frecuentes fueron: talla baja idiopática (TBI) no familiar 32.4%, TBI con retraso constitucional de crecimiento y pubertad (RCCP) 25.4%, TBI familiar 21.4%, talla baja en nacidos pequeños para su edad gestacional (PEG) 13.1%, déficit de hormona de crecimiento 4% y síndrome de Turner 1% (Tabla 1). Los varones fueron diagnosticados más frecuentemente, con una diferencia estadísticamente significativa, de déficit de hormona de crecimiento y de RCCP (p 0.045 y p 0.003 respectivamente) y las mujeres de TBI familiar (p 0.001). El número de visitas medias realizadas fue de 1.4 al año. Recibieron tratamiento con hormona de crecimiento recombinante el 14.5% de los casos. Conclusiones: -La talla baja supone el 30% de los diagnósticos de nuestra consulta. -Los varones son diagnosticados con mayor frecuencia de talla baja. -El 80% de los pacientes fueron diagnosticados de alguna forma de TBI. -Los varones son diagnosticados con más frecuencia de RCCP y déficit de GH y las mujeres de TBI familiar.
P1/d3-025
TALLA FINAL EN PACIENTES CON SÍNDROME DE SILVER-RUSSELL (SSR) EN TRATAMIENTO CON GH A. Vela Desojo(1), N. Portillo Najéra(1), A. Rodriguez Estévez(1), G. Grau Bolado(1), A. Aguayo Calcena(2), I. Rica Etxebarria(2). (1) Hospital Universitario de Cruces,UPV/EHU, Sección de Endocrinología Pediátrica. Barakaldo; (1) Hospital Universitario de Cruces,UPV/EHU /Sección de Endocrinología Pediátrica, CIBERDEM, Barakaldo/ Bizkaia Introducción: Los niños afectos de Síndrome de Silver Russell (SSR) son niños nacidos pequeños para la edad gestacional (PEG). Es un trastorno genético con varios genes implicados, aunque en más de un 60% se debe a una hipometilación de la región 1 del cromosoma 11(Hcr11) y en 10% a una disomía uniparental materna del cromosoma 7 (mDUP7). Además tienen características fenotipicas muy características y otros problemas clínicos. La media de talla final en varones se estima en -3,7 SDS y en mujeres de -4,2 SDS, aunque hay gran variabilidad encontrándose la talla final más afectada en mDUP7. El tratamiento con GH en los PEG está establecido en España desde 2003, sin embargo estos pacientes fueron excluidos en la mayoría de los Comités de GH. Pacientes: Presentamos 4 pacientes afectos de SSR que fueron tratados con GH y que han finalizado el crecimiento (Tabla 1). Comentarios: 1. La talla al final del tratamiento es similar independiente de la mutación que presentan. 2. Desde el inicio de tratamiento hasta el inicio de pubertad la mejoría de talla está entre 1 y 2 SDS. 3. Todos inician pubertad a una edad adecuada. 4. Estos pacientes podían ser candidatos a asociar análogos GnRH al tratamiento con GH en ensayo clínico. 5. A pesar de la talla baja final, todos los casos están satisfechos con el tratamiento.
P1/d3-026
PERFIL DE SEGURIDAD EN EL METABOLISMO HIDROCARBONADO, LIPÍDICO Y TIROIDEO EN NIÑOS TRATADOS CON GH BIOSIMILAR M.J. Rivero Martín, MJ. Alcázar Villar, C. Navarro Moreno, M. Sanz Fernández, P. Pérez Segura Hospital Universitario de Fuenlabrada/ Servicio de Pediatría, Fuenlabrada/ Madrid Introducción: Desde la aprobación por la EMEA del uso de GH biosimilar, el número de pacientes en tratamiento ha ido en aumento. Los ensayos clínicos han demostrado su eficacia y seguridad, pero son necesarios estudios de farmacovigilancia que refrenden su utilización. Método/objetivo: Se lleva a cabo un estudio retrospectivo de niños con déficit de hormona de crecimiento (DHC) y pequeños para edad gestacional (PEG) tratados con GH biosimilar a fin de evaluar el efecto en el metabolismo hidrocarbonado/lipídico y tiroideo. Variables: sexo; peso y talla en diferentes tiempos de estudio (0-3-6-12-24 meses). Parámetros de laboratorio basales y a los 6-12-24 meses: Glucemia, hemoglobina A1C, colesterol, triglicéridos, TSH, y T4L. Se registran otros acontecimientos adversos. Resultados: Se incluyen 18 niños. 4 DGH y 14 PEG (76,5%). Todos los niños con DHC son varones; en los PEG la relación varones/mujeres fue: 4/10. La DE de talla pasó de -3.18 a -2.43, -2.39 y -2 a los 6-12-24 meses (p<0.05) Los niveles medios de IGF-1 fueron: inicio 115.4 ng/ ml y a los 6 meses 250.2 ng/ml (p<0.05). HA1C media pretratamiento era 4.7% y a los 12-24 meses: 4.9 y 5.2 respectivamente (p:ns). 4 pacientes presentaron colesterol entre 200-225 mg/dl (2 pretratamiento), 2 consiguieron control con medidas dietéticas y 2 mantienen CT<225 mg/ dl. El incremento medio de colesterol a los 6-12-24 meses fue: 8.9 mg/dl-2.5 mg/dl-2 mg/dl, respectivamente (p:ns). El incremento medio de triglicéridos a los 6-12-24 meses fue: 12.9-10.7-13.1 mg/dl respectivamente, (p:ns) El incremento medio de TSH a los 6-12-24 meses fue: - 0.14, -0.72 y -1.21 mcU/ml (p:ns). El valor medio de T4 libre pasó de 1.2 ng/dl a 1, 0.8 y 0.9 ng/dl a los 6-12-24 meses respectivamente (p<0.05). En 2 casos se suspendió el tratamiento: uno cambio de dispositivo y otro enfermedad de Perthes bilateral. Conclusiones: El mayor porcentaje de niños con tratamiento con GH son PEG. Hay una mejoría de talla significativa en el tratamiento con GH biosimilar. GH biosimilar ofrece un perfil de seguridad adecuado en cuanto al metabolismo de HC, lipídico y tiroideo a corto-medio plazo. Son precisos estudios de diseño prospectivo y mayor tamaño muestral.
P1/d3-027
ESTUDIO DE EDAD OSEA. VARIABILIDAD INTRA-OBSERVADOR S. De la torre Santos(1), L. Bertholt(2), E. Maldonado Ruiz(1), M.L. Ariza Sánchez(1), M. Cabanillas Boto(1), S. Alberola López(3). (1) Complejo Asistencial de Palencia. Servicio de Pediatría, Palencia; (2) Centro de Salud Aguilar de Campoo. Palencia; (3) Centro de Salud Jardinillos. Palencia Objetivo: Realizar un estudio de evaluación de la variabilidad intra-observador en seis evaluadores en relación con los métodos TW2 y TW3-RUS de Tanner y el método de Greulich y Pyle. Material y metodos: La población está compuesta por un grupo de 150 niños (75 varones y 75 mujeres) a los que se les realizó una radiografía de mano izquierda para el estudio de su edad ósea. Los niños proceden de todo el Área Sanitaria y son atendidos por pediatras. El rango de edades se encuentra entre los 18 meses y los 18 años. El cálculo de la edad ósea ha sido realizado aplicando el sistema TW2 y TW3 RUS de Tanner así como el método de Greulich y Pyle. Se ha realizado un análisis de concordancia mediante el coeficiente Kappa de Cohen ponderado para la comparación de los diferentes estadios de los 13 huesos del método TW2 y TW3 RUS de Tanner. Además se aplicó el cálculo del coeficiente de correlación intraclase (CCI) y análisis de Bland- Altman para los valores de todas las edades óseas. Las valoraciones han sido realizadas por 6 personas diferentes: un médico en formación y dos facultativos experimentados para los métodos de Tanner y otro tanto para el método de Greulich y Pyle. Resultados: Los resultados más relevantes se muestran en la siguiente tabla. Conclusion: La valoración de las edades óseas en la infancia mediante los métodos de Tanner (TW2 y TW3) y Greulich y Pyle obtienen excelentes concordancias clínicas intra-observador en todos los evaluadores.
P1/d3-028
DESENSIBILIZACIÓN A MECASERMINA EN UNA PACIENTE ALÉRGICA CON DÉFICIT PRIMARIO IGF1 I. López Martínez(1), D. Gutiérrez Fernández(2), M. José Anguita(3), J.L. Lechuga Campoy(4), E. Romero Castillo(5), A-M. Lechuga Sancho(6). (1) Complejo Hospitalario Universitario Insular Materno-Infantil de Las Palmas de Gran Canaria, Las Palmas de Gran Canaria; (2) Alergia, Hospital Puerta del Mar, Cádiz; (3) Farmacia clínica, Hospital Puerta del Mar, Cádiz; (4) Pediatría, UCA, Hospital Puerta del Mar, Cádiz; (5) Hospital Puerta del Mar, Cádiz; (6) Pediatría, UCA/UGC Pediatría Hospital Puerta del Mar, Cádiz El crecimiento longitudinal de los pacientes con déficit primario de IGF1 (factor de crecimiento similar a la insulina tipo 1), se ve gravemente afectado, condicionando una talla baja, habitualmente por debajo de 3 DE. El único tratamiento disponible para estos casos es la IGF1 recombinante (mecasermina). Desde que la FDA aprobase el tratamiento en 2005, se han descrito reacciones alérgicas en un 8% de sujetos tratados en diversos ensayos clínicos. Sin embargo solo se han publicado dos casos de alergia en pacientes con deficiencia primaria. Presentamos el caso de una paciente con deficiencia primaria de IGF1 y antecedentes de asma y alergias alimentarias y a ácaros, que desarrolla una reacción alérgica tras la administración de mecasermina, consistente en aparición de lesiones habonosas, primero en piernas y posteriormente en brazos y tórax al décimo día de tratamiento que obligó a suspender el tratamiento temporalmente. Con el fin de poder continuar la terapia, desarrollamos una pauta de desensibilización, con inyecciones subcutáneas en dosis de concentración creciente, llegando a la concentración comercial, 10 mg/ml, y con una separación entre ellas de 15 minutos, hasta alcanzar la dosis de 0.12 mg/kg (dosis máxima recomendada). Una hora antes, la paciente fue premedicada con dexclorfeniramina, deflazacort y montelukast. En la actualidad la paciente ha retomado su tratamiento diario, con buena velocidad de crecimiento y sin notificación de nuevas reacciones alérgicas. Conclusión: El tratamiento con mecasermina puede ser continuado en pacientes con reacción alérgica primaria a ésta, tras someterse a una pauta de desensibilización rápida. Hasta donde sabemos, este el primer caso de desensibilización exitosa a la mecasermina en un paciente con deficiencia primaria de IGF1.
P1/d3-029
ENANISMO PRIMORDIAL OSTEODISPLÁSICO MICROCEFÁLICO. DESCRIPCIÓN DE UN PACIENTE PORTADOR DE UNA NUEVA MUTACIÓN p.Q396X EN EL GEN PCNT (PERICENTRINA) M.A. Guagnelli Martinez, D. Yeste Fernández, M. del Campo Casanellas, I. Valenzuela Palafoll, A. Carrascosa Lezcano Hospital Vall d’Hebron, Barcelona Introducción: El Enanismo Primordial Osteodisplásico Microcefálico tipo II (MOPD II) es la forma más frecuente de enanismo primordial. Sus características incluyen: retraso severo en el crecimiento intrauterino, talla baja proporcionada extrema menor al metro de altura en el adulto, fenotipo facial peculiar con micrognatia y microcefalia severas con cociente intelectual normal-bajo. Pacientes afectos de esta entidad fueron diagnosticados de síndrome de Seckel, pero durante la última década se han identificado 10 genes presentes en diferentes síndromes de enanismo primordial. Entre ellos PCNT, que codifica la pericentrina, una proteína presente en los centrosomas que interviene en la regulación de la mitosis y del ciclo celular. Su ausencia determina un número total menor de células en el organismo. Presentación: Niña de 2 años y 9 meses de edad, peso 5200g (-5.5DE), talla 66 cm (-7.5DE), PC 39 cm (-8.3 DE). Producto de un embarazo a término, peso al nacimiento 1200 g, consanguinidad de los padres, sin otros miembros de la familia afectados. Sus características fenotípicas incluyen facies peculiar con frente amplia, puente nasal amplio con alas nasales delgadas, micrognatia y cubitus varum izquierdo. El perfil bioquímico sanguíneo y la determinación de hormonas tiroideas son normales. IGF-I de 75.3 ng/ ml, IGFBP3 de 2.5 mg/dl. Pico de secreción máxima de GH, 5.4 ng/ml. La serie esquelética muestra huesos largos de forma tubular con ensanchamiento en las epífisis, vértebras aplanadas, coxa vara izquierda. La edad ósea estimada es de 18 meses. La RM de región hipotálamo-hipofisaria es sugestiva de hipoplasia hipofisaria. El estudio genético efectuado mediante secuenciación masiva y el análisis de los genes relacionados con enanismo microcefálico pone de manifiesto la existencia de una mutación en homocigosis en el gen PCNT c.C1186>T que provoca la aparición prematura de un codón de parada p.Q396X. Los padres son portadores de la misma mutación en heterocigosis. Esta mutación no ha sido descrita previamente en la literatura científica. Conclusiones: Las técnicas de secuenciación masiva facilitan la aproximación a las enfermedades con heterogeneidad genética como el enanismo primordial microcefálico. La relación entre las proteínas reguladoras del ciclo celular y estos síndromes puede constituir un nuevo modelo de estudio del desarrollo y crecimiento humano.
P1/d3-030
¿SE PUEDE PREDECIR LA RESPUESTA AL TRATAMIENTO CON HORMONA DE CRECIMIENTO EN NIÑOS CON DÉFICIT DE HORMONA DE CRECIMIENTO IDIOPÁTICO? R. Fernando Martínez, A. Beisti Ortego, A. De Arriba Muñoz, M. Ferrer Lozano, E. Mayayo Dehesa, J.I. Labarta Aizpún Hospital Infantil Universitario Miguel Servet (Servicio de Pediatría), Zaragoza Objetivos: Identificar variables clínicas y analíticas que sirvan como predictores de respuesta en niños con déficit de hormona de crecimiento idiopático (DGHI) tratados con hormona de crecimiento recombinante humana (rhGH). Material y métodos: Estudio retrospectivo de 30 niños con DGHI tratados con rhGH y controlados hasta talla adulta. Se determinaron parámetros perinatales (edad gestacional, longitud, índice ponderal), potencial genético (talla genética, talla media parental), nivel máximo GH, estadio puberal, auxológicos (talla, IMC, edad ósea (EO), velocidad de crecimiento (VC), pronóstico de crecimiento (PC)) y analíticos (IGF-1, IGFBP-3) en el punto inicial, cuarto mes, primer y segundo año de tratamiento, así como talla adulta. Los resultados se expresaron en SDS. Se valoró la respuesta al tratamiento a largo plazo mediante cuatro variables: ganancia de talla adulta SDS respecto a talla inicial SDS (TAdultaSDS-T0SDS) y respecto al PC inicial, talla genética y talla media parental. Se analizaron posibles predictores de respuesta. Resultados: Se observó buena respuesta a corto plazo, con un 46.7%, 73.3% y 83.3% de buenos respondedores, usando como criterios de buena respuesta a los 12 meses la duplicación de VC inicial, aumento VC >2.5 cm/año y aumento VC >1.5SDS, respectivamente. También se constató una buena respuesta a largo plazo, con ganancia media de talla adulta de 1,14 SDS (máximo 3,25 SDS). No se halló correlación estadísticamente significativa entre ganancia de talla (TAdultaSDS-T0SDS) y variables perinatales, sexo, estadio puberal, dosis media, duración del tratamiento o grado DGHI. Se hallaron correlaciones estadísticamente significativas entre ganancia de talla (TAdultaSDS-T0SDS) y los parámetros de buena respuesta a corto plazo (VC, incremento de talla), potencial genético, retraso EO y auxológicos (Tabla). Las cuatro variables de ganancia de talla adulta se correlacionaron negativamente con VC previa al tratamiento (p=0.003 R2=0.274, p=0.042 R2=0.145, p=0.035 R2=0.150, p=0.034 R2=0.150, respectivamente). Conclusiones: La mayor ganancia de talla adulta se observó en los pacientes con mayor retraso EO, mayor potencial genético y mayor retraso de crecimiento previo. Los parámetros auxológicos durante los dos primeros años de terapia demostraron ser los mejores predictores de respuesta final y deberían tenerse en cuenta para optimizar e individualizar la terapia con rhGH en niños con DGHI.
P1/d3-031
LA HORMONA DE CRECIMIENTO ES EFICAZ PERO ¿SIEMPRE NECESARIA? C. Fernández Ramos(1), A. Rodríguez Estévez(2), G. Grau Bolado(2), A. Vela Desojo(2), O. Martínez-Múgica Barbosa(2), I. Rica Echevarria(2) (1) Hospital Universitario Basurto, Endocrinología Infantil, Bilbao, Vizcaya; (2) Endocrinología Infantil. Hospital Universitario Cruces, Baracaldo La experiencia clínica de tratamiento con hormona de crecimiento (GH) en pacientes deficitarios es amplia. Hemos de valorar los resultados que se obtienen con sentido crítico. Objetivo: Analizar la respuesta al tratamiento con GH en pacientes deficitarios que han alcanzado su talla final (TF). Ver si existen subgrupos de pacientes con una respuesta diferente o si hay variables auxológicas que nos determinen la eficacia del tratamiento. Material y métodos: Estudio descriptivo multicéntrico de pacientes deficitarios tratados que finalizaron su crecimiento entre 2004-2012. Hemos estudiado talla-SDS y EO al inicio de la terapia y de la pubertad. Hemos establecido subgrupos en la muestra: Déficit transitorio (T) vs déficit permanente (P) y déficit orgánico (O) vs déficit idiopático (I). Denominamos buena respuesta a una talla final ≥ talla diana. Cálculo de SDS según Estudio Transversal Bilbao 2011. Estudio estadístico con SPSS v17. Resultados: Muestra: 89 pacientes (38% mujeres) procedentes de dos hospitales terciarios. Duración media de terapia: 5,3±2,2 años. El 52% de la muestra fueron maduradores tardíos y el 42% asociaba una talla baja familiar (TBF). En la tabla se describen los subgrupos. Respuesta a GH: La eficacia global es buena: el grupo alcanza su talla diana. Los pacientes que superan la diana (58%) no se diferencian del resto en las variables estudiadas, salvo en una menor talla diana. La mejoría en la talla-SDS durante el tratamiento es progresiva: la talla inicial es estadísticamente inferior a la del comienzo de la pubertad y ésta, inferior a su talla final. Diferencias entre subgrupos: el pico de GH tras estímulo farmacológico fue menor en el grupo O vs I (3,7 vs 5,9 ng/ml; p=0,002). Los subgrupos de I y T, iniciaron la terapia con mayor edad. Entre varones que inician tratamiento después de los 11 años (n: 36), hay mayor proporción de transitorios (89% vs 60% p= 0,046) e idiopáticos (86% vs 52,6% p=0,01) que en el resto. Sin embargo, en relación con la eficacia del tratamiento se comportan como el grupo global. Conclusiones: La respuesta global es satisfactoria. La elevada proporción de déficit transitorio permite que sospechemos que algunos pacientes han sido pubertades tardías.
P1/d3-033
EVALUACIÓN DEL EJE SOMATOTROPO EN UN GRUPO DE PACIENTES CON SÍNDROME DE CORNELIA DE LANGE M.A. Vicente Gabás(1), C. Baquero Montoya(2), M.C. Gil Rodríguez(2), M. Hernández Marcos(2), M.E. Teresa Rodrigo(2), B. Puisac Uriol(2), I. Bueno Martínez(2,3), J. Pié Juste(2), J. Garagorri Otero(3), F. J. Ramos Fuentes(2,3), G. Bueno Lozano(3) (1) Servicio de Pediatría. Hospital Clínico Universitario, Zaragoza; (2) Unidad de Genética Clínica y Genómica Funcional. Departamentos de Farmacología - Fisiología y Pediatría, Facultad de Medicina. Universidad de Zaragoza, Zaragoza; (3) Genética Clínica, Departamento de Pediatría, Hospital Clínico Universitario ¨Lozano Blesa¨, Zaragoza; (4) Endocrinología Pediátrica, Departamento de Pediatría, Hospital Clínico Universitario ¨Lozano Blesa¨, Zaragoza Introducción: El síndrome de Cornelia de Lange (SCdL) es una enfermedad genética rara (OMIM #122470) con afectación multiorgánica que se caracteriza por un fenotipo facial distintivo, retraso del crecimiento pre y postnatal, discapacidad intelectual y malformaciones de extremidades superiores. Objetivo: Valoración del crecimiento y del eje somatotropo en un grupo de pacientes con SCdL. Métodos: Estudio observacional y transversal con datos retrospectivos de 8 pacientes con SCdL (6 niños y 2 niñas, de 2 a 14 años, 7 prepúberes). Se recogieron datos relativos al crecimiento en el nacimiento, a los 4 años y en el momento actual: peso, talla (Carrascosa, 2008), edad ósea (Greulich y Pyle) y talla diana, expresados en desviaciones estándar (DE) para edad y sexo. Los análisis hormonales incluyeron la IGF-1 (ng/mL), IGFBP-3 (μg/mL) y pruebas de valoración de hormona de crecimiento (GH) (estímulo con clonidina y perfil nocturno). Resultados: Al nacer, 6 fueron pequeños para la edad gestacional (PEG), con mayor afectación de longitud que de peso (peso -1,70 DE [-3,46; -0,01], longitud -2,20 DE [-5,2; +0,39]). A los 4 años de vida, dos de los casos presentaron crecimiento recuperador (talla media de -1,9DE [-2,61; -1,19]). En la valoración actual la talla se encontró a -1,89DE, a -1,14DE para su talla diana. El estímulo con clonidina dio una respuesta tras estímulo inferior a 10ng/ml en 6/7 (pico medio de 6,28ng/mL [2,33; 8,99]); el perfil nocturno de secreción de GH mostró cifras <3 ng/ mL/hora en 7/8 pacientes (media de 2,11ng/mL/ hora [1,15; 2,56]). Las cifras de IGF-1 e IGFBP3 no mostraron alteraciones significativas. La edad ósea era patológica en la mitad de los casos, en 3 estaba retrasada y adelantada en el paciente con estudio somatotropo normal. Conclusiones: El grupo de niños con SCdL estudiado ha presentado criterios auxológicos al nacimiento compatibles con ser PEG, con mayor afectación de la longitud que del peso. La mayor parte de ellos no han presentado crecimiento recuperador en los primeros cuatro años de vida. Los datos del estudio de secreción de GH reflejan la presencia de una disfunción neurosecretora que puede contribuir al retraso de crecimiento postnatal de estos niños.
P1/d3-034
ANÁLISIS RETROSPECTIVO DE FACTORES QUE CONDICIONAN UNA BUENA ADHERENCIA AL TRATAMIENTO CON HORMONA DE CRECIMIENTO EN SITUACIONES HABITUALES DE USO. B. Puga González(1), Á. Ferrández Longás(1), E. Mayayo Dehesa(2), J.I Labarta Aizpún(2) (1) Centro Andrea Prader, Zaragoza; Centro Andrea Prader, Zaragoza; (2) Hospital Infantil U. Miguel Servet, Zaragoza; Hospital Infantil U. Miguel Servet, Zaragoza Introducción: La adherencia a cualquier tratamiento es imprescindible para lograr su máxima eficacia, sin olvidar el esfuerzo socio-sanitario que conlleva. Objetivo: Comprobar la adherencia al tratamiento en pacientes tratados con GH. Metodología y casuística: En 2010, 50 familias respondieron a una encuesta sobre: nivel socioeconómico, información recibida, motivación, dispositivo de inyección, dosis perdidas, expectativas y satisfacción con los resultados, etc. En 18 casos (11 mujeres, 7 varones) con una edad de 11,6±2,7 años, 10 con déficit de GH y 8 nacidos PEG por CIR, disponemos de la información proporcionada por un dispositivo electrónico durante 16,3±12,5 meses. Los datos anonimizados fueron obtenidos previo consentimiento informado de los padres. Resultados: En el grupo total hay una pérdida media mensual de 3,3±2,1 inyecciones: 2,4±1,7 (edad 10,5±2,5 años) en los que inyectaban los padres ó 6,4±3,1 en los que se autoinyectaban (14,7±1,6 años). En el 1er grupo la talla ganó 0,29 DS/año vs 0,21 DS/ año antes del registro; en el 2º grupo 0,07 DS/año vs 0,34 DS/año antes del registro. En un pequeño grupo (10,5±3,0 años) el registro del dispositivo desde el inicio del tratamiento, administrado por los padres, la pérdida mensual a lo largo de 2,1±0,5 años fue de 1,4±1,5 dosis. Según la clase social la peor adherencia se observa en la media-alta. El 16,7% admite pérdidas por viajes, fines de semana, vacaciones. La información recibida fue suficiente para el 72,2% y mucha para el 22,2%, desconociendo el 33,3% las consecuencias de las pérdidas. El 72,2% están motivados y satisfechos, aunque el 50% cree que no está creciendo bien. Al 66,7% les gusta el dispositivo por no ver la aguja, ser de fácil manejo y poder comprobar las dosis. Conclusiones: Según muestra el dispositivo electrónico la adherencia al tratamiento es claramente mejorable. Una información inicial exhaustiva es el 1er paso para mejorarla. Durante la pubertad y adolescencia los que se autoinyectan pierden muchas dosis. La personalidad a esas edades, el haber alcanzado una talla aceptable o próxima a la talla adulta, entre otras, explicarían las pérdidas. Los de clase social media baja muestran mejor adherencia.
P1/d3-036
NUEVA MUTACIÓN DEL GEN IGF1R EN UN PACIENTE CON TALLA BAJA Y DÉFICIT DE ATENCIÓN E HIPERACTIVIDAD L. Salamanca Fresno(1), L. Gutiérrez Pascual(1), A. Gómez Núñez(2), M.A. Molina Rodríguez(1), I. González Casado(1), A. Campos Barros(3) (1) Hospital Universitario La Paz / Servicio de Endocrinología Pediátrica, Madrid; (2) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz; Madrid; (3) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz y Centro de Investigación Biomédica en Enfermedades Raras (CIBERER, U753), Instituto Carlos III, Madrid Introducción: El síndrome de resistencia a IGF-I (SR-IGF1) se caracteriza por retraso del crecimiento intrauterino y postnatal con niveles normales o supranormales de IGF-I e IGFBP3, microcefalia acompañada de rasgos dismórficos y déficit intelectual variable. SR-IGF1 está causado por anomalías genómicas o mutaciones que afectan al gen IGF1R (15q26.3). Caso clínico: Se presenta el caso de un paciente remitido a los 10 años por talla baja y estancamiento ponderal. Seguimiento en Neurología desde los 5 años por trastorno por déficit de atención e hiperactividad con impulsividad y agresividad en tratamiento con Metilfenidato. Entre otros antecedentes destacan enuresis nocturna, alergias alimentarias, asma y estreñimiento funcional. Parto postérmino con peso al nacimiento en -1,99 DE y longitud en -2,42 DE. Antecedentes familiares de obesidad e hipotiroidismo con talla diana de 166 + 5 cm. No rasgos dismórficos ni alteraciones a la exploración física con peso en -1,3 DE, talla en -2,01 DE y perímetro cefálico en -2,35 DE. El estudio de talla baja muestra normalidad en perfil tiroideo, estudio de enfermedad celíaca negativo y edad ósea acorde a cronológica. Las somatomedinas muestran valores elevados con IGF1: 387ng/mL (P95; 1,68 DE) e IGFBP3: 5,91 ug/ mL (P89; +1,26 DE). Estudios moleculares: Análisis mutacional de las secuencias codificantes, transiciones intrón/exón y secuencias reguladoras conocidas de IGF1R mediante HRM y secuenciación de las variantes detectadas. Resultados: Se objetiva una mutación puntual de cambio de sentido en heterozigosis no descrita previamente: c.2155C>T, que altera un aminoácido filogenéticamente muy conservado, p.Arg719Cys, localizado en el exón 10 de IGF1R, dominios de Fibronectina III y Tyrosin-protein kinase. La misma ha sido clasificada como probablemente patogénica por las herramientas bioinformáticas Align GVGD, SIFT PolyPhen 2.2 y MutationTaster. Conclusiones: Hemos identificado una nueva mutación de IGF1R en heterozigosis asociada a CIR, déficit del crecimiento postnatal, microcefalia y trastornos de la conducta sin otros rasgos dismórficos, en presencia de niveles supranormales de IGF-I e IGFBP3. Se recomienda el análisis mutacional del gen IGF1R en pacientes con niveles elevados de IGF1 y BP3 asociado a hipocrecimiento pre y postnatal.
P1/d3-037
CARACTERÍSTICAS NEONATALES Y EVOLUCIÓN HASTA LOS CINCO AÑOS DE NIÑOS NACIDOS PEQUEÑOS PARA LA EDAD GESTACIONAL (PEG) CON MENOS DE 1500 GR. C. Luzuriaga Tomás(1), J.L. Guerra Díez(1), T. Galván Luzuriaga(2) (1) Hosp. Univ. Marqués de Valdecilla. Endocrinología Pediátrica, Santander; (2) Fundación Valdecilla. Santander Introducción: Los pacientes pequeños para su edad gestacional (PEG) recuperan crecimiento (90%). Los nacidos con muy bajo peso (<1500 gr) pueden recuperar talla más lentamente o no alcanzar la normalidad. Objetivos: Describir una población de niños PEG menores de 1500 gramos; sin patologías graves neonatales; analizar el aporte calórico e incremento ponderal en los 30 primeros días durante su ingreso; valorar su crecimiento pondoestatural a los 5 años. Material y métodos: Estudio descriptivo retrospectivo de pacientes nacidos entre 2001-2005 con peso inferior a 1500 gramos, clasificados PEG por tablas de referencia. Análisis estadístico (SPSS v15). Resultados: 124 pacientes con peso<1500gr; 27 cumplen criterios PEG (21,77%): 20 niños/7 niñas. Media Edad Gestacional: 31,88±2,24 (28-35 semanas). Crecimiento: Medidos a los 5 años n=19(70,37%): de estos, 4 niños(♂) presentan SDStalla<-2 (21,05%), definidos como no recuperadores; 4 presentan IMC>17 con 5 años, considerado sobrepeso para su edad (Cole et al. 2000). El incremento ponderal medio en los primeros 30 días de vida: 12,60 ± 7,22 gr; (ganancia en peso intrauterino adecuado 15 g/kg/día): Recuperadores:14,41±10,02 vs No Recuperadores:12,11±6,66. Aporte calórico: la media de cal/kg administradas a pacientes No Recuperadores a los 15 y 30 ddv es superior a los Recuperadores (15días de vida: 103,75±27,02vs102,40±26,73; 30días de vida:130, 02±16,08vs116,28±17,9199). Evolución: respecto a los No recuperadores; un paciente inicio tratamiento con GH, a los 6,2 años, aumentando la SDS talla, en 18 meses 0,89, SDS talla actual -1,67 (dosis habituales 0,035 mg/día). Otro paciente asocia patología digestiva y retraso psicomotor leve, SDS en talla (-4,24) con 6 años (no controlado en endocrinología infantil). Los otros 2 son seguidos en atención primaria con recuperación de talla a los 6 años. Conclusiones: 1) Aunque el aporte calórico sea suficiente, no todos los niños realizan una ganancia ponderal adecuada los primeros 30 ddv. La evolución durante el primer mes de vida puede ser clave para establecer sus necesidades futuras 2) Existen pacientes PEG que no alcanzan una talla adecuada tras cinco años, ¿la recuperación en talla de los niños PEG menores de 1500 gr., puede ser más tardía que los PEG no pretérminos con peso >1500 gr.?. 3) A los 5 años ya hay pacientes con sobrepeso.
P1/d3-038
NUEVA MUTACIÓN EN GH1 L. Castro Feijoo(1), C. Quinteiro García(2), J. Barreiro Conde(3), P. Cabanas Rodríguez(3), R. Leis Traba-zo(4), L. Loidi Fernández de Trocóniz(2) M. Pombo Arias(1) (1) Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia, Santiago de Compostela; (2) Fundación Pública Galega de Medicina Xenómica. Santiago de Compostela; (3) Unidad de Endocrinología Pediátrica, Crecimiento y Adolescencia. Universidad Hospital Clínico Universitario de Santiago de Compostela; (4) Unidad de Nutrición Pediátrica. Universidad y Hospital Clínico Universitario de Santiago de Compostela El déficit de GH se define como la combinación de anormalidades auxológicas, clínicas, bioquímicas y metabólicas causadas por la falta o insuficiencia de secreción de GH. El gen GH1 situado en 17q22.24, codifica un polipéptido de 191 aminoácidos y forma parte de un cluster que incluye 5 genes de estructura similar y homología del 92-98%. GH1 es el gen más estudiado en el déficit aislado de la hormona de crecimiento y mutaciones en él han sido detectadas en alrededor del 12,5 y 10% de los casos familiares y esporádicos, respectivamente. Paciente y métodos: Niña de 7,3 años de edad que consultó a los 3,4 años por presentar grave retraso del crecimiento. Producto de un embarazo gemelar dicigótico. Parto a término por cesárea. Peso de RN: 2.360 gr (SDS-2,22). Longitud RN: 43 cm (SDS-3,87). En el periodo neonatal muestra ictericia y episodios de hipoglucemia. Fenotipo sugerente de déficit de GH. También presenta estrabismo convergente. La hermana gemela tiene un crecimiento normal y no se observan rasgos reseñables. Talla Diana: 160,3 cm (SDS: -0,54). Auxología y datos bioquímicos al diagnóstico (ver tabla). Cariotipo: 46XX. RNM de región hipotálamo hipofisaria: Normal. *Análisis genético del gen GH1: amplificación por PCR y secuenciación cíclica de todos los exones, zonas flanqueantes y región promotora proximal. La paciente presenta en el intrón 3, en heterocigosis, la mutación c.291+27G>A, no descrita con anterioridad. El estudio fue realizado también en la hermana y padres que no muestran la alteración encontrada. *Evolución: Se inició tratamiento con hormona de crecimiento a los 3,6 años a una dosis de 0,025 mg/ kg/día. La paciente ha mostrado una mejoría clínica importante con una variación de SDS de talla de: +3,46 (ver tabla). Se corrigió el estrabismo luego de intervención oftalmológica. Conclusión: Se presenta el fenotipo, evolución clínica y respuesta al tratamiento con GH en una paciente con una mutación de novo en el gen GH1, c.291+27G>A, que afecta el procesamiento del mRNA debido a que está localizada en un enhancer de splicing intrónico y que se describe, por primera vez, como causa de Déficit de GH tipo II.
P1/d3-039
HIPOCRECIMIENTO POSTNATAL EN PACIENTE CON MUTACIÓN EN HOMOCIGOSIS DE IGFALS L. Ruiz Pérez(1), M. Zapico Álvarez-Cascos(1), A. Campos Barros(2) (1) Unidad de Endocrinología infantil. Hospital General Universitario de Alicante, Alicante; (2)Instituto de Genética Médica y Molecular. Hospital Universitario La Paz. Madrid Introducción: Los pacientes con deficiencia primaria de IGF-1 presentan un déficit del crecimiento postnatal asociado a niveles muy disminuidos de IGF-1 e IGFBP3, en presencia de niveles normales o elevados de GH. Estudios recientes muestran que mutaciones del gen codificante de ALS (IGFALS) representan un defecto molecular frecuente en dichos pacientes, lo que indica que ALS desempeña una función importante en la regulación del crecimiento postnatal. Caso clínico: Niño de 4 años que consulta por talla baja. Antecedentes personales: Recién nacido a término con peso adecuado a edad gestacional (peso: 2670 kg). Antecedentes familiares: Talla paterna 168 cm; talla materna: 156,6 cm. Talla diana: 168,8 cm. Exploración física: Peso: 12,3 Kg (DE: -2,15), Talla: 89,9 cm (DE: -3,84). Velocidad de crecimiento: 3,4 cm/ año (DE: -3,38). Pruebas complementarias: Edad ósea: 3 años y medio; IGF-1 < 10 ng/ml, IGFBP-3 < 0,5 mcg/ml, test estimulación de GH normal, test de generación de IGF-1 (IGF1 basal < 10 ng/ml, IGF1 tras prueba < 10 ng/ml). Estudio molecular: Mutación en homocigosis en el gen IGFALS, codificante de la subunidad ácido lábil. Madre: portadora en heterocigosis de la misma mutación. Padre (desconocido): no realizado estudio genético. Hermano: normal. Ante el diagnóstico de déficit primario de IGF-1, se inicia tratamiento con IGF-1 presentando buena respuesta clínica y analítica. Conclusiones: La mutación del gen codificante de ALS (IGFALS) representa un factor etiológico en el hipocrecimiento postnatal asociado al déficit importante de IGF-1. Estos pacientes se pueden beneficiar con el tratamiento con IGF-1, requiriendo un estrecho seguimiento clínico por el riesgo de presentar hipoglucemias y otros efectos secundarios (cardiovasculares, renales).
P1/d3-040
EFICACIA Y SEGURIDAD DEL TRATAMIENTO CON HORMONA DEL CRECIMIENTO EN NIÑOS PEQUEÑOS PARA SU EDAD GESTACIONAL: ANÁLISIS INTERMEDIO A 3 AÑOS. ESTUDIO MULTICÉNTRICO J.P. López Siguero(1), M.J. Martínez-Aedo(2), J.A. Bermúdez(3), N. Cabrinety(4), J.L. Lechuga(5), A. Gutiérrez(6), F. Macias(7); M. Núñez(8); J. Bosch(9), R. Espino(10); R. Torralva(11). (1) Hospital Materno-Infantil, Málaga; (2) Hospital Materno-Infantil de Málaga; (3) Hospital Virgen Macarena de Sevilla; (4) Hospital Sagrado Corazón de Barcelona; (5) Hospital Virgen del Mar, Cádiz; (6) Hospital de Murcia; (7) Hospital de Jerez; (8) Hospital Materno-Infantil, Badajoz; (9) H. Arnau de Vilanova; (10) H Virgen de Valme, Sevilla; (11) Merck, Madrid Se presentan los resultados de eficacia y seguridad del análisis intermedio a tres años del estudio multicéntrico observacional (SGA 27143) de una cohorte de niños pequeños para su edad gestacional (PEG) tratados con hormona del crecimiento (GH). Objetivos: Primario: Estudiar cambios en la sensibilidad a la insulina (HOMA) que pudiesen asociarse a un riesgo de desarrollo de diabetes tipo II, junto a otros factores de síndrome metabólico. Secundarios: Describir los cambios auxológicos tras tratamiento con GH. Métodos: Estudio observacional, no aleatorizado, abierto, de una cohorte de niños PEG tratados con GH con criterios de inclusión del Ministerio de Sanidad. Sujetos: 299 sujetos recibieron al menos una dosis de GH (población de seguridad) (54% varones). 272 de ellos continúan en seguimiento, y 147 han cumplido al menos un año de tratamiento. El tratamiento comenzó a una edad media de 7.4 (2,8) años. Variables: Datos demográficos: edad cronológica y sexo. Datos auxológicos: peso y altura basal, altura y peso el primer, segundo y tercer año, edad ósea, velocidad de crecimiento y HOMA. Análisis estadístico: Descriptivo de variables expresadas como media (SDS) y desviación estándar entre años. Resultados: Ver tabla. No se encontró relación entre el HOMA y el resto de variables estudiadas (p<0,05). Ver Tabla I Seguridad: 5 pacientes (1,7%) tuvieron AEs: diabetes tipo II (retirada del estudio), ansiedad (retirada), tiroiditis, molestias en el lugar de inyección y aumento de transaminasas. Conclusiones: El tratamiento con GH estimuló el crecimiento en niños PEG (ganancia promedio de 1,36 SDS) sin avance significativo de edad ósea. El tratamiento mostró un buen perfil de seguridad. A pesar del incremento del HOMA el primer año, no se consideró clínicamente relevante. La posterior tendencia del HOMA no fue estadísticamente significativa respecto al valor del primer año. |
| References |