
| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):194-198 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.188 |
| Gónadas |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
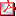 Tabla - P1/d3-078 Tabla - P1/d3-078 |
| Tabla - P1/d3-079 |
|
|
P1/d3-074
QUISTE DE OVARIO FUNCIONANTE CON IMPORTANTE REPERCUSIÓN CLÍNICA QUE PRECISA RESECCIÓN QUIRÚRGICA A. Díaz Moro(1), S. Gautreaux Minaya(1), L. Regueras Santos(1), R. Quiroga González(1), G. Lorenzo(2), F. Manrique(1) (1) Complejo Asistencial Universitario de León, León; (2) Complejo Asistencial Universitario de Burgos, Burgos Introducción: Los quistes ováricos son lesiones que se presentan más frecuentemente durante la edad pediátrica y forman parte de la etiología de la pubertad precoz periférica PPP. Las lesiones ováricas, aunque raras, abarcan un amplio espectro de patologías desde los quistes funcionales hasta tumores malignos. Caso clínico: Niña de 2 años y 5 meses remitida a la consulta de endocrinología infantil por telarquia bilateral de aparición a los 2 años que asocia sangrado vaginal en el último mes durante 4 días. Antecedente de telarquia a los 6 meses que regresó espontáneamente en 1-2 meses. En la exploración física se objetiva Tanner II (T2, P1, Aa). Edad ósea de 2 años y 6 meses. En el hemograma se le detectó una discreta anemia ferropénica; un perfil hormonal (TSH, T4 Libre, Prolactina, DHEAS, Androstendiona, cortisol) normal, cariotipo 46 XX; Test de Procrin sin signos de activación del eje hipotálamo-hipofisario gonadal (FSH a las 3 horas 0,603 miliU/ml, LH a las 3 horas: 0,304miliU/ml, 17 beta-estradiol a las 24 horas: 44,7pcg/ml); en la ecografía abdomino-pélvica se objetiva una imagen quística en ovario derecho de 3,5 cm., marcadores tumorales negativos. Clínicamente presenta posteriormente persistencia de sangrados vaginales mensuales de 4 días de duración, durante 6 meses, con incremento de telarquia (Tanner 3). Se le realizan ecografías seriadas objetivándose a los seis meses aumento de la lesión a 4,5 cm con aspecto hipoecoica bien delimitada. Por los hallazgos ecográficos y la clínica se decide tratamiento quirúrgico, en la que se conserva el ovario derecho; tras la intervención no vuelve a presentar nuevos episodios de sangrado. Comentarios: En general, el tratamiento de los quistes ováricos pediátricos no es quirúrgico, precisando controles ecográficos. Dentro de los posibles diagnósticos diferenciales estarían los tumores ováricos. En nuestro caso, la repercusión clínica y el gran tamaño nos hizo inclinarnos por el tratamiento quirúrgico. Hay que hacer hincapié en lo importante que es la conservación del ovario.
P1/d3-075
REVISIÓN DEL MANEJO DEL QUISTE OVÁRICO EN UN HOSPITAL DE TERCER NIVEL EN LOS ÚLTIMOS 10 AÑOS J. Bote Mohedano, O. Patiño Hernández, AC. Barreda, MA. Molina, P. Olivares, I. González Casado Hospital General Universitario La Paz, Madrid
Introducción: Los quistes ováricos son lesiones mayoritariamente benignas. Ocasionalmente son hormonalmente activos. Su diagnóstico se realiza mediante ecografía. Su visualización requiere un diagnóstico diferencial con tumor ovárico para evitar ooforectomías innecesarias. Puede no visualizarse si realizamos el estudio fuera del momento oportuno. Material y métodos: Estudio descriptivo retrospectivo del manejo del quiste ovárico en un centro hospitalario de tercer nivel en lo últimos 10 años (N=9 pacientes con diagnóstico confirmado). Resultados: De los 9 casos en 2 se realizó diagnóstico intraútero, confirmándose al nacimiento. Ninguno presentó signos de pubertad precoz. Ambos tenían parámetros analíticos normales. Se realizó cirugía en uno de ellos con anatomía patológica (AP) de quiste simple (QS). Ambos casos tuvieron evolución favorable. En los 7 casos restantes la edad media al diagnóstico fué de 6.0 ± 3.83 años. Motivo principal de consulta: telarquia II-III 71,5% (57,1% con hiperpigmentación areolar), 3/7 sangrado vaginal, pubarquia II 3/7 y axilarquia positiva 1/7. El 71,5% tenía edad ósea adelantada. Ningún caso presentaba síntomas de alteración del eje suprarrenal, ni manchas café con leche. Coagulación normal en el 100% de los casos. Elevación del estradiol y marcadores tumorales (α-FP, beta-HCG) positivos en 5/7 casos. Todos los casos presentaban valores prepuberales de LH-FSH y normalidad en Prolactina y perfil tiroideo. ECO pélvica: útero puberal 5/7 y prepuberal 2/7; quistes sin signos de malignidad (QS) 3/7, quistes complicados (QC) 4/7. De los QS se intervino 1 con resultado AP: quiste simple benigno. De los 4 QC se intervienen 2 con AP: 1 quiste simple benigno, el otro aún pendiente de resultado. Los 2 QC manejados con actitud expectante: al mes negativizan marcadores tumorales si fueron positivos, normalizan estradiol y desaparece imagen en ECO. Conclusiones: Los QS son principalmente benignos. Los signos de malignidad ecográficos son: presencia de calcificaciones, septos, imagen de contenido heterogéneo y sin tendencia a la resolución. Ante un diagnóstico de certeza de QS, por su tendencia natural a la remisión, el manejo debe ser conservador.
P1/d3-076
CISTOADENOMA OVÁRICO CON CISTOADENOMA HEPÁTICO EN LA EDAD PEDIÁTRICA J.A. Nieto Cuartero, D. Azorín Cuadrillero, S. Gallego Chacón, A. Castillo Robleda, J. Asensio Antón, S. Vila Duplá Hospital Infantil Universitario Niño Jesús. Endocrinología, Madrid; Introducción: Presentamos la coexistencia de un cistoadenoma hepático con un cistoadenoma del ovario derecho de 63 x 49 x 57 mm. En una revisión de 240 casos recogidos en el Hospital de Niños de Philadelphia EE.UU. (Journal of Pediatric Surgery, Vol. 38, N.3, 2003. pp 331-335), 123 en 51,2% eran de naturaleza no neoplásica. Los pacientes con tumores epiteliales tenían una edad media 13,9 ± 4 años. El diagnóstico histopatológico incluía 9 cistoadenomas serosos (49%) y 3 cistoadenomas mucinosos (16%). Presentamos por su rareza en la edad pediátrica, una niña operada con 7 años de edad el 5 de Noviembre de 2007 de un cistoadenoma hepático Materiales y métodos / Objetivos: Niña en la actualidad de 11 años y 2 meses con talla de 1,576 m peso de 47,7 Kg edad ósea de 12 años y edad talla de 13 años y 6 meses. Tanner III, aún no menarquia. La paciente fue operada de cistoadenoma hepático con extirpación de tumoración de la cara posterior del lóbulo hepático derecho de 10 x 10 x 12 cm, que presentaba tres pequeños quistes que fueron vaciados. En la actualidad presenta una tumoración en ovario derecho compatible con cistoadenoma ovárico derecho. Datos complementarios: hemograma con vsg normal. Bioquímica sérica: normal. Orina: normal. Estudio hormonal: LH 089 mU/ml y 5,6 mU/ml. Estradiol 69 pg/dl. Estudio tiroideo: normal. Progesterona: 2,1 ng/ml. IGF I, IGFBP-3 , insulina y HOMA: Normal. Anticuerpos antitiroideos y marcadores de celiaquía: negativos. Marcadores tumorales βHGC α-fetoproteina, CEA y CA 19,9 fueron normales. No se realizó inhibina plasmática preoperatoria y pendiente resultados CA 125. Resultados: Destacar la buena evolución en nuestra paciente de su patrón de desarrollo así como la coexistencia de cistoadenoma hepático y ovárico en una paciente pediátrica. Conclusiones: 1. La coexistencia en una paciente de cistoadenoma ovárico y cistoadenoma hepático es excepcional en la edad pediátrica. 2. Se debe de vigilar la aparición de posibles cistoadenomas de otra localización a lo largo de su existencia así como el riesgo de adenocarcinoma ovárico.
P1/d3-077
PUBERTAD PRECOZ POR TUMOR DE CÉLULAS GERMINALES MEDIASTÍNICO EN UN PACIENTE CON SINDROME DE KLINEFELTER E. Puerto Carranza, M. Caimari Jaume, D. de Sotto Esteban, J.A. Salinas Sanz, L. Solaeche Fernández Hospital Universitari Son Espases, Palma de Mallorca Introducción: El síndrome de Klinefelter (SK) es una anomalía de la diferenciación sexual con una prevalencia estimada de 1/600 varones. La mayoría de pacientes presentan hipogonadismo, siendo la edad puberal la más frecuente al diagnóstico. La incidencia de tumores no está aumentada salvo el riesgo de tumor de células germinales mediastínico (TCGM), que es 50 veces mayor. Presentamos el caso de un paciente con SK y pubertad precoz de progreso rápido producida por TCG mediastínico. Caso clínico: Varón de 8 años y 10 meses que acude remitido por pubertad precoz de 4 meses de duración. Presenta antecedentes de rápido aumento del tamaño del pene y caracteres sexuales secundarios junto con la aparición de acné. A la exploración física destaca: 138 cm (+0,78DE), 32,5 kg(+0.04 DE) e IMC 17.07 (-0.26DE). A1P4G2 y volumen testicular 4 ml. En el estudio analítico se objetivan FSH y LH suprimidas con testosterona de 11,5 ng/ml y marcadores tumorales elevados: AFP 44,76 ng/ml y BHCG 25,2 mUI/ ml. Ante la sospecha de pubertad precoz de origen tumoral, se realizan RMN cerebral y ecografía abdominal, que son normales. La ecografía testicular evidencia microlitiasis y el TAC torácico una masa mediastínica anterior de 54 mm por lo que se procede a toracoscopia y resección macroscópica completa del tumor. El estudio anatomopatológico confirma el diagnóstico de teratoma inmaduro grado I con un microfoco de tumor del seno endodérmico. Tras la resección, se normalizan los marcadores tumorales. Posteriormente, se realiza quimioterapia según protocolo MAKEI 96 (PEI) y se encuentra en remisión completa a los 6 meses de finalizar el tratamiento. Conclusiones: La pubertad precoz en pacientes varones debe hacer sospechar origen tumoral. En nuestro paciente con SK, caracterizado por desarrollo puberal tardío y mayor riesgo de presentar TCGM, la sospecha debe ser aún mayor.
P1/d3-078
MUTACIÓN EN EL GEN NR5A1 (SF1) EN 4 PACIENTES 46,XY. VARIABILIDAD EN FENOTIPO A. Rodriguez Estévez(1), G. Pérez de Nanclares Leal(1), I. Costa Alcacer(2), J. Haro Mora(3), R. Fernández G-Salazar(3), L. Castaño González(4) (1) Hospital Universitario de Cruces, Barakaldo; (2) Hospital de Manises (Valencia); (3) UTIG-HRU Carlos Haya (Malaga); (4) Hospital Universitario de Cruces-UPV/EHU-CIBERER (Barakaldo) Introducción: SF-1/NR5A1/Ad4BP (MIM184757) es un receptor nuclear que regula multiples genes implicados en el desarrollo adrenal y gonadal, la esteroidogénesis y reproducción. Presenta un amplio espectro de fenotipos en 46XY, desde la disgenesia gonadal completa (DGC) con/sin insuficiencia adrenal al hipospadias escrotal. Casos: 4 sujetos 46XY, 2 con asignación de sexo femenino y 2 masculino. Ninguno asocia insuficiencia adrenal. Todos con mutación en heterocigosis. Diagnóstico tardio en las mujeres (C1 y C2) por ausencia de pubertad y amenorrea primaria. Los 2 varones presentan cuadro de Alteración de la Diferenciacion Sexual (ADS) con hipospadias escrotal y criptorquidia asociado en uno de ellos a micropene. En el caso 2 con niveles de Beta hCG 57,68 mU/ml, no se objetiva gonadoblastoma. En las 2 mujeres el estudio del gen SRY fue normal. Se realiza analisis mediante PCR y secuenciación directa el gen NR5A1. El potencial efecto patogénico de las mutaciones previamente no descritas se analizo in silico usando los software SIFT (http:// sift.jcvi.org/) y Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/). Conclusiones: Las mutaciones en NR5A1 son frecuentes en pacientes 46,XY con ADS de diferente severidad. La mayoria presenta mutaciones en heterocigosis, y no asocian insuficiencia adrenal.
P1/d3-079
INDUCCIÓN DE LA PUBERTAD CON ESTROGENOTERAPIA TRANSDÉRMICA EN NIÑAS CON HIPOGONADISMO J.L. Gómez Llorente, M.A. López Sánchez, J. Momblan de Cabo, A. Bonillo Perales Hospital Torrecárdenas, Almería Introducción: Actualmente se está promoviendo el uso o ventajas del tratamiento con estrógenos transdérmicos para la indución de la pubertad en niñas con hipogonadismo frente a la estrogenoterapia oral, debido a su eficacia comparable y a una menor aparición de efectos secundarios. Objetivo: Evaluar el desarrollo de caracteres sexuales secundarios y crecimiento tras tratamiento de inducción con estrógenos transdérmicos en niñas con hipogonadismo. Material y métodos: Estudio retrospectivo descriptivo. Se incluyen todas las niñas con hipogonadismo que han iniciado el tratamiento de inducción de la pubertad con estrógenos transdérmicos, desde Junio de 1989 hasta diciembre de 2012. Variables de estudio: Edad, talla, peso, desarrollo puberal (estadios Tanner), dosis de estrogenoterapia, tratamiento con hormona de crecimiento, velocidad de crecimiento, Densidad mineral ósea, crecimiento uterino, alteración perfil hepático (transaminasas). Resultados: 8 niñas fueron incluidas en el estudio (de ellas 7 completaron el tratamiento con estrógenos y progesterona). Seis con síndrome de Turner, una con hipogonadismo postradioterapia y otra con hipogonadismo postquirúrgico tras ovariectomía. La edad media de inicio del tratamiento con estradiol en parches fue de 13,38 años con edad ósea media de 12,31 años. La media de meses de tratamiento con estradiol hasta alcanzar la telarquia completa fue de 27,14 meses. La edad media de aparición de la menarquia fue a los 15,4 años habiendo transcurrido una media de 19,14 meses desde el inicio de la terapia con estrógenos. La talla media en el síndrome de Turner fue de 147 cm y en el resto de las pacientes de 158 cm. Se realizó ecografía pelvica en el 71,4% de las pacientes, presentando todas ellas estrogenización completa. La densitometría ósea se lleva a cabo en el 62,5% de las pacientes, presentando cifras osteopénicas una de ellas. En ninguna de nuestras pacientes aparecen alteraciones de las transaminasas. Conclusiones: La pauta de estrogenoterapia transdérmica en nuestro centro ha conseguido una evolución del desarrollo puberal adecuada con aparición de la menarquia en tiempo esperable, datos comparables en eficacia a los estudios realizados con estrogenoterapia oral. No se han producido alteraciones en el perfil hepático y se ha alcanzado una mineralización ósea normal.
P1/d3-080
QUISTES OVÁRICOS PERINATALES : ACTITUD TERAPÉUTICA P. Terradez Marco(1), I. Güemes Heras(1), R. Fornes Vivas(1), A. Cuñat Romero(2), A. Plasencia Couchud(3), M. Sanchis Plasencia(4) (1) Hospital Casa de Salud / Servicio de Pediatria, Valencia; (2) Hospital Casa de Salud. Valencia; (3) Hospital Casa de Salud Radiodiagnóstico. Valencia; Hospital Virgen del Consuelo. Valencia; (4) Centro de Salud de Cheste. Cheste Introduccion: Los quistes ováricos (NOC), aparecen en el 0,039% recién nacidos vivos (RNV), representando el tumor abdominal más frecuente a esta edad (30%). Su etiopatogénia no está definida, pudiendo estar implicada la estimulación hormonal. Se clasifican, según Nussbaum en simples (anecoicos, homogéneos y de paredes finas) y complejos (con ecos en su interior, septos, restos hemorrágicos, detritus). Según su tamaño en pequeños (< 20 mm) o grandes (>20 mm). La mayoría son unilaterales, benignos e involucionan antes de los 24 meses. Suelen ser asintomáticos. La complicación mas frecuente es la torsión ovárica. Material y métodos: Análisis de estudio retrospectivo de historias clínicas de recién nacidos, dados de alta con el diagnóstico de NOC (CIE-9: 620.2) en la última década. Resultados: Se diagnosticaron 9 NOC, incidencia 1/2.728 RNV (0,036%) Detección prenatal entre la semana 28 y 34 de gestación en todos los casos. Ninguna gestación cursó con diabetes materna, toxemia, hidramnios o isoinmunización Rh y los fetos no presentaron malformaciones asociadas. Nacidos entre la semana 37 y 40 de gestación, por cesárea en el 33%, siendo sólo una electiva por perímetro abdominal fetal aumentado. Del total de NOC, 4 fueron simples y 5 complejos. Solo hubo un caso bilateral. El ovario más afectado fue el izquierdo (62,5%). Todos los casos simples y 2 complejos se resolvieron espontáneamente antes del segundo año de vida. De los tres NOC complejos tratados, en 2 se hizo ooforectomia tras torsión ovárica y en uno quistectomia laparoscópica por crecimiento progresivo (mayor a 5 cm), pudiéndose preservar el ovario. El estudio anatomopatológico confirmó el diagnóstico descartando signos histológicos de malignidad. Conclusiones: La tendencia natural de los NOC es la involución espontánea, sobre todo si son simples. La complicación más frecuente es la torsión ovárica, sobre todo en los complejos, que conlleva la ooforectomia. En los NOC mayores de 5 cm, existe controversia entre establecer tratamiento conservador o quirúrgico de entrada para evitar la torsión ovárica. Consideramos como primera opción el seguimiento clínico y ecográfico, indicando la quistectomía laparoscópica en los casos que presenten complicaciones o excesivo crecimiento, con el fin de preservar el ovario. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites