
 No English version available for this Article No English version available for this Article |
|||||||||
| Rev Esp Endocrinol Pediatr 2013;4(1):48-53 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2012.Sep.86 | |||||||||
| Diagnóstico de Síndrome de Kallmann en varones durante el periodo neonatal: revisión de los últimos 15 años | |||||||||
| Kallmann's syndrome diagnosis in males at birth: review of the last 15 years | |||||||||
| Sent for review: 19 Jan. 2012 | Accepted: 4 Sep. 2012 | Published: 16 Sep. 2013 | |||||||||
| Ariadna Campos Martorell, Laia Vega Puyal, María Clemente León, Diego Yeste Fernandez, Maria Angeles Albisu Aparicio, Antonio Carrascosa Lezcano | |||||||||
| Unidad de Endocrinología Pediátrica. Departamento de Pediatría. Hospital Universitari Vall d'Hebron (UAB). Barcelona | |||||||||
| Correspondence:Ariadna Campos Martorell, Unidad de Endocrinología Pediátrica. Departamento de Pediatría, Hospital Universitari Vall d'Hebron (UAB), Barcelona E-mail: arcampos@vhebron.net | |||||||||
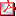 Tabla 1 - Características de los pacientes diagnosticados de SK en los primeros meses de vida Tabla 1 - Características de los pacientes diagnosticados de SK en los primeros meses de vida | |||||||||
| |||||||||
Diagnóstico de Síndrome de Kallmann en varones durante los primeros meses de vida: revisión de los últimos 15 años.
Introducción El Síndrome de Kallmann (SK) se ha definido clásicamente como la asociación de hipogonadismo hipogonadotropo con alteraciones olfatorias como anosmia o hiposmia. Esta asociación fue establecida inicialmente por el patólogo español, Maestre de San Juan en 1856, cuando realizando una autopsia a un paciente masculino asoció la presencia de genitales externos subdesarrollados con la ausencia de bulbos olfatorios. Kallmann en 1944 describió el trastorno como un síndrome genético basándose en los informes clínicos de 11 pacientes de tres familias con eunucoidismo y anosmia. En 1963 Morsier y Gauthier sugirieron la disfunción hipotalámica como la etiología del síndrome. Las características principales del SK son el hipogonadismo hipogonadotropo y la anosmia, la fisiopatología de los cuales radica en el defecto de migración, desde su origen común, de las neuronas de la hormona liberadora de gonadotropina (GnRH) y de las neuronas olfativas durante el desarrollo embrionario. El hipogonadismo hipogonadotropo es un trastorno del eje hipotálamo-hipófisis-gonadal que resulta de una insuficiente secreción de GnRH, produciendo una inadecuada gametogénesis y una insuficiente producción de esteroides sexuales. Aunque un micropene o una criptorquidia nos podrían hacer pensar en el diagnóstico de un hipogonadismo hipogonadotropo en el periodo neonatal, en la mayoría de ocasiones el diagnóstico de SK se realiza durante la adolescencia por un retraso en el desarrollo de los caracteres sexuales secundarios1. Además de las principales características del SK como el hipogonadismo hipogonadotropo y la anosmia, en el SK se pueden asociar otras muchas anomalías. Las más frecuentes son pérdida de audición, agenesia renal, sincinesia (el movimiento involuntario de una mano cuando la otra se mueve, o movimiento en espejo), anomalías de línea media y disfunción cerebelosa. Con menos frecuencia se han asociado al SK anomalías músculo-esqueléticas, anomalías óculomotoras y cardiopatías. Como muchas de estas anomalías ya están presentes en el momento del nacimiento, nos ofrecen una buena oportunidad para el diagnóstico precoz1. La incidencia del SK es muy superior en el sexo masculino, estimándose de 1 de cada 8.000-10.000 varones, y de 1 de cada 40.000- 50.000 mujeres2,3. El patrón de herencia es variable, habiéndose descrito casos de herencia ligada al X, herencia autosómica dominante y herencia autosómica recesiva. Actualmente se conocen siete genes que se han relacionado con el SK (KAL1, FGFR1, PROK2, PROKR2, FGF8, NELF y CHD7), sin embargo, sólo en el 30% de los pacientes con diagnóstico clínico se llega a encontrar una mutación en uno de estos genes4. El gen KAL-1 (Xp 22.3), responsable del SK ligado al X, es el más frecuentemente detectado, en un 10-20% de los casos. KAL-1 codifica para una glicoproteína de la matriz extracelular denominada anosmina-1 que favorece la adhesión celular de las neuronas. En ausencia de dicha proteína no existe el “andamiaje” para la migración de las neuronas productoras de GnRH desde la placoda olfativa hasta el hipotálamo4. El gen denominado KAL-2 (8p11.2-12) o FGRF1 se ha detectado en otro 10% de los pacientes diagnosticados de SK. El resto de genes implicados PROK2, PROKR2, FGF8, NELF y CHD7 son los responsables del SK en el restante 10%4. El diagnóstico precoz del SK es importante porque puede ofrecerse tratamiento para normalizar el tamaño del pene desde los primeros meses de vida y realizar consejo genético precoz a la familia. El objetivo de este trabajo es analizar las características clínicas y bioquímicas de cuatro pacientes con diagnóstico de SK durante los primeros cuatro meses de vida, la clave diagnóstica en todos ellos ha sido la presencia de micropene. Todos ellos presentan anomalías asociadas al SK. Materiales y métodos Se realizó la revisión sistemática de las historias clínicas con diagnóstico clínico de SK durante los primeros meses de vida durante los últimos 15 años en un hospital de tercer nivel. Se recogieron las variables clínicas (edad, sexo, hallazgos fenotípicos de sospecha), analíticas (niveles de gonadotropinas y de testosterona), al diagnóstico así como las resultantes tras el test de betaHCG. Del mismo modo, registramos los resultados de las pruebas de imagen (ecografía abdominal, RNM craneal), y resultados de la audiometría y del estudio genético en los casos en los que se realizó, con el fin de caracterizar cada caso. Resultados Se encontraron 4 casos, todos ellos varones. En los cuatro, fue la evidencia clínica del micropene lo que llevó a la sospecha diagnóstica de SK. La edad media al diagnóstico fue de 70 días de vida (entre 34–128 días). Para el diagnóstico del hipogonadismo hipogonadotropo se realizó la determinación hormonal basal y tras el test de beta-HCG (500 U días alternos) en sus dos modalidades, el test corto (3 dosis) en sólo uno de los pacientes, y el test largo (6 dosis) en los otros 3 pacientes. En tres de los pacientes se hizo el estudio del gen KAL-1 mediante secuenciación tras amplificación por PCR de los 14 exones del gen, en la Unidad de Medicina Molecular- FPGMX del Hospital Clínico Universitario de Santiago de Compostela, España. Los resultados de los pacientes se resumen en la tabla 1. Los datos clínicos al diagnóstico fueron micropene en los cuatro pacientes, criptorquidia unilateral en uno y criptorquidia bilateral en dos, dos de ellos presentaban además testes pequeños (< 1 ml). La determinación hormonal basal se realizó entre los 30 y los 115 días vida. Los cuatro pacientes presentaban testosterona <20 ng/dl, LH <0,38 U/L y FSH<2,2UI/L. En tres de los pacientes se realizó el test largo de beta-HCG (6 dosis) en los cuales se obtuvo un valor de testosterona intermedia entre 160 y 247ng/dl y un valor de testosterona final entre 441y 1078,7ng/dl. El test corto de beta-HCG de 3 dosis se realizó en un paciente, con un valor de testosterona final de 66 ng/dl. El tamaño del pene aumentó hasta un valor normal (>2,5cm) en los 3 pacientes que realizaron el test largo, no fue así en el que se realizó el test corto. A todos los pacientes se les practicó una resonancia magnética cerebral. Todos ellos tenían el área hipotálamo-hipofisaria sin alteraciones y ausencia de tractos y/o bulbos olfatorios. En uno de ellos se descubrió además una hipoplasia del vermis cerebeloso. Se detectó una agenesia renal izquierda en la ecografía abdominal en uno de los pacientes. En los tres primeros pacientes, debido a su corta edad, la existencia de anosmia es desconocida, en el paciente número 4 que actualmente tiene 15 años sí que se ha confirmado hiposmia. En tres pacientes se realizó el estudio del gen KAL-1 (Xp22.3), encontrándose mutaciones no descritas previamente en dos de ellos, el estudio del tercer paciente resultó normal. Se ha realizado estudio audiométrico al paciente número 1, a la edad de 4 años, que muestra una hipoacusia izquierda grave de predominio neurosensorial, sin poder descartarse afectación del componente de transmisión. El estudio audiométrico de los pacientes 2 y 3 está pendiente en el momento de la redacción de este artículo. El paciente de más edad, no recibió ningún tratamiento hasta la pubertad, cuando se iniciaron inyecciones intramusculares de testosterona cada 3 semanas, en dosis progresivas. DISCUSIÓN El diagnóstico del SK es poco frecuente durante los primeros meses de vida, diagnosticándose habitualmente durante la adolescencia por falta de desarrollo puberal. Sin embargo, es importante estar atentos a la presencia de anomalías genitales en el sexo masculino, como son el micropene y unos testículos pequeños o criptorquídicos para sospechar la existencia de hipogonadismo hipogonadotropo que deberá ser tratado precozmente. Aunque clásicamente el SK se ha caracterizado por la presencia de hipogonadismo hipogonadotropo y alteraciones olfatorias, también se ha asociado con otras anomalías, muchas de las cuales pueden pasar desapercibidas si no se piensa en ellas y no se realizan las exploraciones complementarias pertinentes para diagnosticarlas. Ante la sospecha clínica de un SK debe practicarse una analítica hormonal basal, en la que unos niveles normales o disminuidos de gonadotrofinas y testosterona nos orientan hacia el diagnóstico de hipogonadismo hipogonadotropo. Normalmente, en el sexo masculino, los niveles de testosterona y gonadotrofinas presentan un aumento de secreción durante los primeros seis meses de vida, para posteriormente disminuir y permanecer en niveles muy bajos hasta la pubertad, momento en que se pone en marcha de nuevo el eje hipotálamo-hipofisario gonadal. El diagnóstico de hipogonadismo hipogonadotropo puede confirmarse con un test de HCG (hormona coriónica gonadotrópica), hormona con estructura similar a la LH, que estimula las células de Leydig testiculares incrementando la síntesis de testosterona. Así este test tiene una finalidad diagnóstica y terapéutica pues también se suele observar aumento del tamaño del pene y del volumen testicular, pigmentación de la zona genital y descenso de los testes a la bolsa escrotal. Existen dos variantes del test de HCG, el test corto de 3 días y el test largo de 6 días. A tres de los pacientes se les practicó el test largo, que nos confirmó la sospecha de hipogonadismo hipogonadotropo y también se produjo un aumento del tamaño del pene en los tres pacientes. Los valores de testosterona no aumentaron lo suficiente con el test corto de 3 días y tampoco se observaron cambios clínicos en el pene del paciente al que se le practicó dicho test, por lo que en nuestra experiencia, si bien limitada, recomendaríamos preferentemente la realización de un test largo ante la sospecha de SK, tanto para el diagnóstico como para el tratamiento de estos pacientes. Actualmente la beta HCG ha sido retirada del mercado pero se dispone de una forma recombinante que se puede usar con los mismos efectos. La RMN craneal para el estudio de la región hipotálamo-hipofisaria, y el territorio de los bulbos olfatorios, debe ser practicada ante la sospecha de SK. Cabe decir, que aunque en los 4 pacientes se detectó una alteración en los bulbos olfatorios, esta anomalía puede no estar presente en todos los casos de SK. El estudio genético en el SK es importante para poder dar consejo genético a la familia y al paciente, pero sólo en el 30% de los pacientes se detecta una mutación. El gen KAL-1 (Xp 22.3), responsable del SK ligado al X, es el más frecuentemente detectado, hasta en un 10-20% de los casos, según las series. El segundo en frecuencia con un 10%, es FGRF1, y el resto de genes implicados PROK2, PROKR2, FGF8, NELF y CHD7 son los responsables del SK en el 10% restante4. En nuestra serie, se realizó el estudio del gen KAL-1 (Xp22.3) en tres pacientes, encontrándose mutaciones no previamente descritas en dos de ellos. En el primer paciente se encontró la mutación c.98G>C (p. Arg33Pro) que afecta a un aminoácido poco conservado y lo sustituye por otro con moderadas diferencias fisicoquímicas. Queda pendiente el estudio genético de la madre para averiguar si la mutación se ha producido de novo o ha sido heredada. El estudio del gen KAL-1 del segundo paciente resultó normal. En el tercer paciente se halló la mutación c.1449+2T>A, de la que se desconoce su efecto. Esta mutación consiste en la sustitución de una T por una A en el intrón 10, sin embargo, la predicción teórica de su efecto indica que destruye el sitio dador de splicing, lo cual daría como resultado un ARNm anormal que sugeriría la alteración del fenotipo de este paciente. En la bibliografía, hay pocos estudios de diagnóstico de SK en los primeros meses de vida. En una de las series más largas, estudiada por Abujbara et al en 2004 con 26 pacientes, la presencia de micropene (definido por una longitud inferior a 2.5 desviaciones estándar para la edad y etnia correspondientes) se detecta en un 65% de los individuos con SK y la criptorquidia en el 75% de los pacientes varones, la mayoría de las veces bilateral5. Que la presencia de micropene y criptorquidia sea del 100% en nuestra corta serie es lógico, pues fueron los signos guía que nos hicieron sospechar el SK. La anomalía renal más frecuentemente asociada al SK es la agenesia renal unilateral derecha, cuando en pacientes sin SK es más frecuente del lado izquierdo. Se han descrito otras anomalías renales asociadas como el divertículo renal, el riñón en herradura, el riñón poliquístico y el reflujo vesicoureteral5. El gen KAL-1 es el único que se ha relacionado con las anomalías renales, de hecho, la probabilidad de encontrar una mutación en el gen KAL-1 aumenta hasta el 85% si el paciente presenta agenesia renal6. A todos los pacientes con sospecha de SK se les debe practicar una ecografía renal y si en esta se detectan anomalías, el estudio del gen KAL-1 debería ser el primero en realizarse. En nuestra serie de 4 pacientes hemos encontrado un paciente con agenesia renal izquierda y otro con un reflujo vesicoureteral, en ambos pacientes se ha detectado una mutación en el gen KAL-1. Las anomalías en línea media como agenesia dental o paladar hendido se han relacionado con otros genes como FGFR1, FGF8, y CHD7, por lo que parece razonable testar primero estos genes cuando se detectan este tipo de anomalías. En nuestros pacientes no hemos observado anomalías de línea media7. La pérdida de audición se ha descrito en el 28-40% de los individuos, dependiendo de la series1. La hipoacusia puede ser de conducción y/o sensorial, unilateral en la mayoría de pacientes, aunque también se han descrito casos de forma bilateral. Los problemas auditivos se han relacionado con mutaciones en los genes KAL1, FGFR1,FGF8, PROKR2, y CHD7. A todo paciente con la sospecha de SK se debería practicar una valoración audiométrica. Se ha realizado estudio audiométrico al paciente número 1 que muestra una hipoacusia izquierda grave de predominio neurosensorial, sin poder descartarse afectación del componente de transmisión. En resumen, el SK incluye otras anomalías a parte de las históricamente descritas como el hipogonadismo hipogonadotropo y la anosmia. La presencia de micropene o criptorquidia nos debe hacer sospechar un hipogonadismo hipogonadotropo e investigar otras posibles anomalías asociadas. Para el diagnóstico del SK se debería practicar una analítica hormonal basal y otra tras estímulo con HCG, preferiblemente, en base a nuestra experiencia, el test largo con 6 dosis. A todo paciente con sospecha de SK se le debería practicar una resonancia magnética cerebral que incluya el territorio de los bulbos olfatorios, una ecografía abdominal para buscar posibles alteraciones renales y un examen audiométrico. En los pacientes con anomalías renales y alteraciones audiométricas se debería comenzar el estudio genético por el gen KAL-1. Reconocer estas anomalías durante el periodo neonatal y la época escolar es importante para un correcto diagnóstico clínico y molecular del SK y un tratamiento eficaz del micropene y otras anomalías asociadas, así como poder ofrecer consejo genético a la familia.
Conflictos de interés Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo. | |||||||||
| References | |||||||||
1.Kaplan JD, Kwan A, Bernstein JA, Hudgins L. Clues to an early diagnosis of Kallmann syndrome. Am J Med Genet Part A 2010;152A:2796-801. 2.Cadman SM, Kim SH, Hu Y, Gonzalez-Martinez D, Bouloux PM. Molecular pathogenesis of Kallmann’s syndrome. Horm Res 2007;67:231-242.[Pubmed] 3. Kulkarni ML, Balaji MD, Kulkarni AM, Sushanth S, Kulkarni BM. Kallmann’s syndrome. Indian J Pediatr 2007;74:1113-1115.[Pubmed] 4. Semple RK, Topaloglu AK. The recent genetics of hypogonadotrophic hypogonadism – novel insights and new questions. Clin Endocrinol 2010;72: 427-435. 5. Abujbara MA, Hamamy HA, Jarrah NS, Shegem NS, Ajlouni KM. Clinical and inheritance profiles of Kallmann syndrome in Jordan. Reprod Health 2004;1:5.[Pubmed] 6. Georgopoulos NA, Koika V, Galli-Tsinopoulou A, Spiliotis BE, Adonakis G, Keramida MK, Sgourou A, Koufogiannis KD, Papachatzopoulou A, Papavassiliou AG, Kourounis G, Vagenakis GA. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil Steril 2007;88:1311-1317.[Pubmed] 7. Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, Minowada S, Shimotsuka A, Shishiba Y, Yokozawa M, Yasuda T, Nagasaki K, Hasegawa D, Hasegawa Y, Tachibana K, Naiki Y, Horikawa R, Tanaka T, Ogata T. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab 2004;89:1079-1088.[Pubmed] | |||||||||
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites