
| Rev Esp Endocrinol Pediatr 2012;3 Suppl(1):101-107 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2012.Apr.110 |
| Tiroides |
| Sent for review: 13 Apr. 2012 | Accepted: 13 Apr. 2012 | Published: 30 Apr. 2012 |
| Congreso SEEP |
| Correspondence:Congreso SEEP |
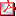 Tabla - O3d3-022 Tabla - O3d3-022 |
| Tabla - O3d3-024 |
|
|
O3d3-018 CARACTERIZACIÓN MOLECULAR COMPLETA DEL GEN THRB (ISOFORMAS THRB-1 Y THRB-2) EN PACIENTES CON SÍNDROME DE RESISTENCIA A HORMONAS TIROIDEAS N. Rico Ríos (1), A. Reche Martínez (1), M. A. Molina (1), C. Álvarez Escolá (1), B. Lecumberri (1), M.P. de Miguel (2), B. Roldán (3), J. C. Moreno (1) (1) Hospital Universitario La Paz, Madrid, (2) Hospital Clínico San Carlos, Madrid, (3) Hospital Universitario Gregorio Marañón, Madrid Introducción: El síndrome de resistencia a hormonas tiroideas clásico (RHT) se debe a alteraciones genéticas en los exones 7-10 del gen THRBeta. Sin embargo existe un porcentaje sustancial (20%) de pacientes con RHT sin defectos en este gen cuya base molecular es desconocida. El gen THRBeta tiene 2 isoformas, una de expresión general (THRB-1) y otra específicamente hipofisaria (THRB-2), que codifica un exón ausente en THRB-1 escasamente estudiado. Objetivo: Estudio genético de la zona codificante completa del gen THRB (isoformas 1 y 2) en una amplia cohorte nacional de pacientes con sospecha de RHT. Pacientes y métodos: Estudiamos a una cohorte de 30 individuos (70% mujeres y 30% hombres) de 28 familias independientes, con 50% de casos índice pediátricos y 50% adultos. PCR y secuenciación directa de exones 3-10 del gen THRB-1 y del exón específico de la isoforma THRB-2 (hipofisaria). Resultados: En niños y adultos respectivamente, la edad media al diagnóstico fue de 5,8 años y 40 años; la TSH media fue de 7.84 mUI/mL (SD=5.33) y 13.81mUI/mL (SD=21,4), (N: 0,27-4,20 mUI/mL); la media de T4L resultó ser 2,41 ng/dL (SD=1.04) y 2,14 ng/dL (SD=1,6) (N: 0,93-1,70 ng/dL) ; y la T3L (fue 1.71 ng/mL (SD=0,76) y 2.15 ng/mL (SD=1,5) (N=0,80-2 ng/mL). Se observó un 20% TDAH, 15% de microadenomas hipofisarios, 30% retraso de la edad ósea, 36% taquicardia, 15% enfermedad de Graves, 35% bocio, 31% autoinmunidad positiva, algunos pacientes presentaron trastornos respiratorios, reflujo, diarrea e hiperquinesia. Se hallaron 12 mutaciones, 9 en casos índice pediátricos: R243Q y R243W (2 casos) en el exón 7; R338W en el exón 9; P453T, P453R (2casos), R438H y M442V en el exón 10) y 3 en adultos (R438H). No se encontraron mutaciones en los exones 3-6 ni en el específico de THRB2. Conclusiones: En nuestra serie, un 50% de pacientes tiene RHT clásica con mutaciones en el gen TRHB. Tras estudiar toda la zona codificante queda sin diagnosticar un porcentaje de casos en los que sería interesante estudiar las zonas UTR, microRNA relacionados y otros genes que codifican cofactores de otros factores de transcripción que coadyuvan la función de TRHB.
O3d3-019 NUEVA MUTACIÓN DEL GEN NKX2-1 CON AFECTACIÓN TIROIDEA, NEUROLÓGICA Y GRAVE ENFISEMA PULMONAR L. Garzón Lorenzo (1), C. M. Moya (2), C. Luna (3), R. Simón (4), M.A. Zaballos (5), P. Santisteban (5), E. Gallego (1), J.C. Moreno (2) (1) Sección de Endocrinología Pediátrica, Hospital Universitario Doce de Octubre, Madrid, (2) Laboratorio de Tiroides, INGEMM-Instituto de Genética Médica y Molecular, Hospital Universitario La Paz, Madrid, (3) Sección de Neumología y Alergia Pediátricas, Hospital Universitario Doce de Octubre, Madrid, (4) Sección de Neuropediatría, Hospital Universitario Doce de Octubre, Madrid, (5) Departamento de Fisiopatología Endocrina y del Sistema Nervioso, Instituto de Investigaciones Biomédicas, CSIC, Madrid Introducción: NKX2-1 es un factor de transcripción que se expresa en tiroides, ganglios basales cerebrales y pulmón. Defectos en este gen causan hipotiroidismo primario, desórdenes neurológicos y distrés respiratorio, pero con gran variabilidad en la presencia y/o severidad de estos fenotipos. Recientemente se ha sugerido que distintos dominios de la proteína NKX2-1 podrían llevar a cabo funciones diferentes en el desarrollo. TAZ es un co-activador de NKX2-1 con gran expresión en tejido pulmonar. Objetivo: Identificar la causa genética y los mecanismos patogénicos de un Síndrome Cerebro-Pulmón-Tiroides en un niño con gravísimo enfisema pulmonar. Paciente y Métodos: Varón de 14 años con hipotiroidismo congénito diagnosticado en screening neonatal (TSH 224µU/ml; T4L 0.6ng/dl) con tiroides in situ. Desde el nacimiento, grave dificultad respiratoria que evoluciona a broncopatía crónica dependiente de oxigenoterapia y corticoterapia. TAC torácico con áreas de destrucción pulmonar. Biopsia con extenso enfisema, fibrosis intersticial y alteración bronquiolar. Presenta además movimientos coreicos continuos desde los 9 meses asociados a retraso psicomotor. PCR y secuenciación del gen NKX2-1. Mutagénesis del plásmido de expresión de NKX2-1. Transfección transitoria de células eucariotas con 3 vectores de expresión: NKX2-1 salvaje, con una mutación amino-terminal (224insG) y otra carboxi-terminal (825delC) junto a vectores reporteros de promotores tiroideos, pulmonares y cerebrales (TG, SP-B, Lhx6) asociados o no al plásmido de expresión TAZ. Ensayos luciferasa, western-blot y microscopía confocal. Resultados: Se identificó una nueva mutación heterocigota de novo en NKX2-1 (c.224insG; p.V75fsX408), la más amino-terminal descrita. La proteína mutante tiene vida media y localización nuclear normales. Sin embargo, no posee capacidad para activar la transcripción de los genes diana de tiroides, pulmón y ganglios basales. En contraste con otra mutación (NKX2-1 c.825delC) descrita en pacientes sin problema pulmonar, la mutación encontrada en este paciente (c.224insG) es incapaz de activar el promotor pulmonar del surfactante pulmonar B incluso en cotransfección con el co-activador pulmonar TAZ. Conclusión: Los estudios moleculares confirman que la mutación c.224insG es la causante del Síndrome NKX2-1 en el paciente. Además sugieren que mutaciones que alteren la capacidad de interacción entre NKX2-1 y TAZ cursaran con problemas pulmonares graves en adición al fenotipo neurológico y tiroideo.
O3d3-020 BASES MOLECULARES DE LOS DEFECTOS EN LA SÍNTESIS DE TIROGLOBULINA C. Villalba Castaño (1), C.M. Moya (2), M.A. Molina (3), M. Polak (4), I. González (3), J.C. Moreno (2) (1) Servicio de Pediatría, Hospital Virgen de la Salud, Toledo, (2) Laboratorio Molecular de Tiroides, Instituto de Genética Médica y Medicina Molecular (INGEMM), Hospital Universitario La Paz, Madrid, (3) Servicio de Pediatría, Hospital Universitario La Paz, Madrid, (4) Endocrinología Pediátrica, Servicio de Pediatría, Hospital des Enfants Malades, Paris Introducción: Los defectos en la síntesis de la Tiroglobulina (DST) (8q24.2-24.3) constituyen una causa relativamente frecuente de dishormogénesis tiroidea. El gran tamaño del gen hace difícil su estudio molecular y requiere una apropiada selección de los pacientes a estudiar basada en la conjunción de Tiroglobulina sérica baja y herencia recesiva. Objetivo: Identificar las claves clínicas, bioquímicas y el defecto genético de un grupo de pacientes con sospecha de DST. Pacientes/Métodos: Estudiamos una cohorte española con hipotiroidismo congénito y TG sérica baja. El estudio molecular se realizó mediante secuenciación de los exones 7, 8, 17, 22 y 38 (mayor frecuencia de mutaciones en caucásicos), MLPA (Multplex Ligand Probe Amplification) y CGH-Arrays (Hibridación Genómica Comparada, ThyroArray® de 23 genes tiroideos, incluido Tiroglobulina). Resultados: Estudiamos 4 pacientes (3 mujeres, 2 varones). La edad media al diagnóstico fue de 3,05±5,3 días. Todos los pacientes, menos uno, presentaban hipotiroidismo congénito con valores medios de TSH de 275±309,5 mU/ml y T4L 1,34±1,39 ng/ml, tiroglobulina de 0,19±0,12 ng/dl y anticuerpos antitiroideos negativos en todos. Dos pacientes presentaban bocio: uno fetal, que precisó L-T4 intraamniótica y otro neonatal. Estos últimos tenían antecedentes familiares de bocio multinodular. En ninguna familia había consanguinidad. Sorprendentemente, una paciente tenía un test de descarga de perclorato positivo (42%). Otra paciente asociaba una exostosis múltiple de herencia paterna (gen EXT1, cromosoma 8q24.11). En el estudio molecular se encontraron mutaciones bialélicas en heterocigosis compuesta en 2 pacientes: 1. p.R77X/p.R321Q, 2. p.R277X/D2585-S2620del (9,8Kb que deleciona el exón 45). En una paciente se encuentra una mutación: p.R277X/_ Conclusiones: La presencia de mutaciones en 3 de 4 pacientes apoyaría la estrategia selectiva de screening molecular. Habría que incluir el CGH-Arrays en el estudio de DST, ya que detecta deleciones inadvertidas en la secuenciación. La mutación más frecuente en la serie (p.R277X) lo es también a nivel mundial. Las mutaciones que encontramos son de herencia paterna. Presentamos un cambio de secuencia no descrito en el exón 8 cuya patogenicidad habrá que demosntrar. La asociación de hipotiroidismo congénito y exostosis múltiple no se ha descrito y podría deberse a una deleción amplia del cromosoma 8.
O3d3-021 APROXIMACIÓN AL DIAGNÓSTICO ETIOLÓGICO DE LAS DISHORMONOGENESIS S. Grau Montero, L. Velásquez Puentes, M. A. Albisu Aparicio, A. Campos Martorell, D. Yeste Fernández, M. Clemente León Hospital Universitario Vall dHebron, Barcelona La dishormonogénesis representa un 10-20% de los hipotiroidismos congénitos. Los trastornos moleculares se sitúan a nivel de receptor de TSH, transportador de yodo (NIS), organificación del yodo (tiroperoxidasa, DOUX2, DUOXA2), síntesis de tiroglobulina y actividad de las desyodasas. Su herencia es autosómica recesiva. Objetivo: Orientar la etiología del defecto de la hormonogénesis en los pacientes con hipotiroidismo congénito y presencia de tiroides ortotópico. Pacientes y métodos: estudio descriptivo en el momento de la reevaluación(r) mediante determinación de TSH, T4L y Tiroglobulina, gammagrafía con I123 y test de descarga de perclorato (TDP) tras tres semanas de suspensión del tratamiento, de los pacientes que presentan tiroides ortotópico y han sido diagnosticados mediante el screening(s) neonatal desde 1990 en Cataluña. Resultados: Identificamos 50 pacientes. Se realizan 43 test de perclorato, a la edad media de 6,09años (rango 2,6–15), de ellos 53,5% eran varones y 46,5% mujeres. Se obtuvieron: 19 test con resultado positivo: 9 casos de posible déficit de tiroperoxidasa(TPO), 3 casos de posible déficit de tiroglobulina(TG), 7 casos de hipertirotropinemia (posible DUOX o déficit parcial de tiroperoxidasa(TPOp); 24 test con resultado negativo: 3 posibles déficits de tiroglobulina, 7 casos de hipotiroidismo transitorio, 11 casos de hipertirotropinemia, 1 sin catalogar, 1 caso con ecografía normal y gammagrafía no captante (posible mutación NIS) y una confirmación diagnóstica de hemiagenesia. Otro caso se diagnosticó de déficit tiroglobulina sin TDP. Conclusiones: La reevaluación diagnóstica mediante determinación analítica y la gammagrafía con test de descarga de perclorato permite orientar el diagnóstico etiológico de los pacientes afectos de dishormonogénesis, previo al estudio genético.
O3d3-022 PRESENCIA DE ANTICUERPOS ANTI CÉLULAS PARIETALES GÁSTRICAS EN PACIENTES CON TIROIDITIS AUTOINMUNE M. Bonet Alcaina (1), S. Ortigosa Gomez (1), P. Ruiz-Cuevas Garcia (2), M. Murillo Valles (3), R. Corripio Collado (4), M. Torrabías Rodas (5), C. Sánchez Garre (6), M.V. Borrás Pérez (7) (1) Hospital Parc Salut Mar, Barcelona, (2) Clínica Girona, Girona, (3) Hospital Universitari Germans Trias i Pujol, Badalona, (4) Hospital de Sabadell, Coorporació Universitaria Parc Taulí, UAB. Sabadell, (5) Hospital General de Vic, Vic, (6) Hospital Mutua de Terrassa, Terrassa, (7) Hospital General de Granollers, Granollers Introducción: En la infancia, a diferencia del adulto, la asociación entre gastritis autoinmune (GA), y enfermedad tiroidea autoinmune (ETAI) está poco caracterizada. Los anticuerpos contra las células parietales gástricas (ACPG) constituyen el principal marcador inmunológico de GA. Hay autores que aconsejan el estudio de ACPG al diagnóstico de ETAI y en caso de positividad determinación anual de gastrina como marcador bioquímico de GA. La hipergastrinemia establece la indicación de endoscopia digestiva alta para valoración histológica. Objetivos:
Material y métodos: Estudio multicéntrico de 49 pacientes afectos de ETAI: 32 con tiroiditis de Hashimoto (25 mujeres, 7 varones con un rango de edad 8-17 años) y 17 con enfermedad de Graves (14 mujeres, 3 varones con un rango de edad 6-19 años). Se determinó ACPG, TSH, T4 libre y anticuerpos tiroideos en todos y en 30 gastrina. Se practicó gastroscopia en un paciente con ACPG positivos e hipergastrinemia. Se evaluó edad al diagnóstico de ETAI, tiempo de evolución de la enfermedad, presencia de otras enfermedades autoinmunes y clínica asociada. Resultados: Se detectaron ACPG positivos en tres pacientes mujeres (2 adolescentes de 19 y 13 años con enfermedad de Graves y 1 de 16 años afecta de tiroiditis de Hashimoto). En dos se determinó gastrina que estaba elevada aunque también se objetivó hipergastrinemia, a un título más bajo, en dos pacientes con enfermedad de Graves y ACPG negativos. La gastroscopia realizada confirmó gastritis atrófica. Un paciente con ACPG positivos presentó anemia ferropénica. Conclusiones:
O3d3-023 REPERCUSION DE LOS NIVELES DE YODURIA DURANTE LA GESTACION SOBRE EL COCIENTE INTELECTUAL EN LA INFANCIA A. Aguayo Calcena (1), G. Grau Bolado (1), A. Vela Desojo (1), A. Aniel-Quiroga (2), M. Espada Sáez (3), G. Miranda Rodríguez (4), Y. Duque Franco ( 4), P. Martul Tobio (1), L. Castaño González (5), I. Rica Etxebarria (1) (1) Sección de Endocrinología Pediátrica, (2) Servicio de Bioquímica-Laboratorio de Hormona, (4) Psicología Sanitaria, (5) Unidad de Investigación, Hospital Universitario de Cruces, Barakaldo, Bizkaia, (3) Laboratorio Normativo de Salud Pública, Departamento de Sanidad, Gobierno Vasco. Introducción: Diversos estudios refieren que los niños en situación de deficiencia de yodo (DY) tienen un menor cociente intelectual, alteraciones del aprendizaje y escasa motivación, pero se desconoce qué nivel de déficit puede empezar a afectar al desarrollo psicomotor. Objetivos: Valorar la relación entre los niveles de yodo en el embarazo y el posterior cociente intelectual (CI) de los niños. Determinar el nivel de DY en las embarazadas susceptible de afectar a su descendencia. Pacientes y métodos: La población total fue de 2246 mujeres gestantes. Se analizaron yoduria, función tiroidea y anticuerpos antiTPO en el primer (1T) y segundo trimestre (2T). Se consideraron seis subgrupos deficitarios de yodo (≤ 150 µg/L). Entre los 6 y 8 años de edad se valoró el CI mediante la escala de inteligencia Wechsler para niños (WISC-IV) en 310 de los nacidos. Se consideró como grupo control a 51 niños cuyas madres mantuvieron yodurias > 150 µg/L, T4L > p10 y antiTPO negativos en ambos trimestres. Resultados: No se encontró correlación entre la yoduria de las embarazadas y el CI de los niños (r-0,023 en 1T y 0,039 en 2T). No se observaron diferencias en los grupos considerados como yodo deficientes respecto a los controles. Al considerar a los niños con CI en los extremos [CI>110 (n=122) o CI<90 (n=32)] no se hallaron diferencias en la medias de las yodurias. Conclusión: En nuestra muestra de niños entre los 6-8 años, no se observaron diferencias con el coeficiente intelectual en relación con los niveles de yodurias durante la gestación. Ayudas: Dpto. Sanidad, Fundación Salud 2000 y Premio José Igea
O3d3-024 HIPOTIROXINEMIA MATERNA EN EL EMBARAZO Y REPERCUSIÓN EN EL DESARROLLO INTELECTUAL DE SU DESCENDENCIA G. Grau Bolado (1), A. Aguayo Calcena (1), A. Vela Desojo (1), A. Aniel-Quiroga (2), M. Espada Sáez (3), G. Miranda Rodríguez (4), Y. Duque Franco ( 4), P. Martul Tobio (1), L. Castaño González (5), I. Rica Etxebarria (1) (1) Sección de Endocrinología Pediátrica, (2) Servicio de Bioquímica-Laboratorio de Hormona, (4) Psicología Sanitaria, (5) Unidad de Investigación, Hospital Universitario de Cruces, Barakaldo, Bizkaia, (3) Laboratorio Normativo de Salud Pública, Departamento de Sanidad, Gobierno Vasco Introducción: Se ha considerado que la hipotiroxinemia materna (HM) en el primer trimestre (1T) del embarazo puede afectar al desarrollo mental en la descendencia, aún con cifras de TSH normales. La positividad en el embarazo de los antiTPO también se ha referido como predictor para un peor desarrollo neurológico. Objetivos: Valorar si existe correlación entre los niveles de T4L en embarazadas y el cociente intelectual (CI) de sus hijos. Evaluar si la HM en primer o segundo trimestre (2T) de embarazo determina un menor CI en la descendencia. Comprobar si la presencia de antiTPO en las madres se relaciona con un menor desarrollo intelectual. Material y métodos: En 2246 mujeres gestantes se analizaron yoduria, función tiroidea (T4L y TSH) y anticuerpos antiTPO en 1T y 2T. Se consideraron seis subgrupos yododeficientes (yodurias ≤ 150 µg/L). Se determinó el percentil 10 para la T4L (1,1 ng/dl en 1T y 0,93 en 2T). Entre los 6 y 8 años de edad se valoró el CI (WISC-IV) en 310 de los nacidos. Grupo control: 51 niños cuyas madres mantuvieron yodurias > 150 µg/L, T4L>p10 y antiTPO negativos en ambos trimestres. Resultados: No se encontró correlación entre la HM y el CI de los niños (r: -0,087 en 1T y 0,016 en 2T). Las medias de CI de los nacidos de madres con T4L110 (n=122) o CI <90 (n=32)] no hubo diferencias en relación a la T4L materna en ninguno de los trimestres. Los antiTPO positivos en las embarazadas (n=44) no determinaron un menor CI en sus hijos. Conclusiones
Ayudas: Dpto. Sanidad, Fundación Salud 2000 y Beca José Igea
O3d3-025 CARACTERIZACIÓN CLÍNICA Y MOLECULAR DE PACIENTES CON HIPOTIROIDISMO CONGÉNITO CENTRAL M. García González (1), M.A. Molina (1), R. Barrio (2), A.C. Barreda (1), B. Lecumberri (1), J. Guerrero (1), I. González (1), J.C. Moreno (3) (1) Endocrinología Pediátrica Hospital Universitario La Paz, Madrid, (2) Endocrinología Pediátrica Hospital Ramón y Cajal, Madrid, (3) Laboratorio Endocrinología Molecular de Tiroides (Instituto de Genética Médica y Molecular), Hospital Universitario La Paz, Madrid El hipotiroidismo congénito central (HCC) no es detectado por cribado neonatal con TSH, y estos pacientes son diagnosticados a posteriori, por sospecha de hipotiroidismo clínico. El HCC incluye defectos hipotalámicos e hipofisarios que el test de TRH puede ayudar a diferenciar. Aunque existen algunos casos de HCC con defectos de TSHB y TRHR, y en diferentes factores de transcripción hipofisarios, la base genética del HCC es fundamentalmente desconocida. Objetivo: Caracterización clínica y estudio molecular en una cohorte española de pacientes pediátricos y adultos con hipotiroidismo congénito central. Pacientes y Métodos: Estudiamos una serie de 14 pacientes (5 varones y 9 mujeres) con edad media al diagnóstico de 7 años (3 m-12 años). Se definió CCH como presencia de T4L disminuida y TSH baja o inapropiadamente baja para la T4L. Con test de TRH largo (180´), se clasificó CCH hipofisario con pico neto <15 mU/L, e hipotalámico con pico de >15 mU/L y/o ratios 30´/0´>4, sin retorno a valores basales (ratio 180´/0´>1.5) (criterios Van Tijn). PCR y secuenciación directa de los genes TRH, TRHR, TSHB, POU1F1 e IGSF1, y arrays-CGH. Resultados: Un 28.6% de los pacientes se clasificaron de HCC hipofisario (TSH de 1.28±1,53mU/L y T4L de 0.6±0.26ng/dl), un 21.4% de HCC hipotalámico (TSH de 2.41± 0.74 mU/L y de T4L de 0.93±0.26 ng/dl), y el resto de HCC indeterminado. 7/14 pacientes presentaban S. Shapiro, Axenfeld-Rieger, Alstrom, Marfan y McCune-Albright. Se identificó una mutación en homocigosis en el gen POU1F1 (p.R265W) en una niña de tres años con los ejes somatotropo (GH basal <0.05), tirotropo (TSH basal <0.005mU/L) y lactotropo (PRL: 0.6ng/ml) afectos. También una deleción total del recientemente identificado gen IgSF1 (Immunoglobulin Superfamily 1) en un paciente con HCC hipofisario y macroorquidismo. Conclusiones: El HCC hipofisario es más intenso que el hipotalámico, por sus diferencias significativamente inferiores de T4L y TSH. Hemos identificado la causa genética del 14% (2/14) de nuestra serie (PO1F1, IGSF1), pero la mayor parte de la base genética del HCC permanece desconocida. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites