
| Rev Esp Endocrinol Pediatr 2012;3 Suppl(1):119-126 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2012.Apr.112 |
| Pósters exposición oral |
| Sent for review: 16 Apr. 2012 | Accepted: 16 Apr. 2012 | Published: 30 Apr. 2012 |
| Congreso SEEP |
| Correspondence:Congreso SEEP |
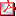 Tabla - MP1d2-001 Tabla - MP1d2-001 |
| Tabla - MP1d2-002 |
| Tabla - MP1d2-008 |
| Tabla 1 - MP1d2-004 |
| Tabla 1 - MP1d2-006 |
| Tabla 1 - MP2d3-010 |
| Tabla 2 - MP1d2-004 |
|
|
MP1d2-001 CARACTERÍSTICAS CLÍNICAS Y ANALÍTICAS DE NIÑOS CON HIPERCOLESTEROLEMIA SEVERA. ESTUDIO GENÉTICO Y ECOGRÁFICO A NIVEL DE GROSOR ÍNTIMA-MEDIA CAROTÍDEO M.L. Bertholt (1), M. Pelaz Esteban (2), I. Palenzuela Revuelta (1), J.L. Guerra Diez (1), M.C. Luzuriaga Tomás (1) (1) Unidad de Endocrinología Pediátrica, (2) Servicio de Radiología Pediátrica Hospital Universitario Marqués de Valdecilla, Santander Objetivo: describir las características clínicas y analíticas de los pacientes con hipercolesterolemia de alto riesgo de los últimos años. Valorar el análisis genético, si la historia clínica es sugestiva. Valorar la repercusión en las arterias (grosor del complejo íntima-media carotideo), medido ecográficamente. Material y Métodos: análisis descriptivo retrospectivo de historias clínicas de pacientes diagnosticados de hipercolesterolemia. Estudio genético del gen del receptor en sangre de LDL (LDLR), y medición con ecografía en modo B, Doppler color y espectral del complejo íntima media en la pared posterior de la arteria carótida común, aproximadamente a 1-2cm de la bifurcación, con sonda lineal de alta frecuencia (12MHz). Resultados: analizamos 27 historias: 51,9% varones y 48,1% mujeres. El 74,1% consultó por hipercolesterolemia. Dos pacientes adoptados 7,4% y el resto el 85,2% tienen antecedentes de primer grado, el 77,8% también de segundo grado, y de tercer grado 18,5% (aunque no bien precisado por falta de datos). El 29,6% contaba con antecedentes de enfermedad cardiovascular severa y/o muerte súbita precoz. El 48,1% presentó comorbilidad (obesidad, hipotiroidismo, talla baja, pseudohipoparatiroidismo). El 29,6% presentó un índice de masa corporal elevado (>2SDS). Edad media al diagnóstico 7,1±3 años. Colesterol total máximo 281,1±35.62mg/dl. LDL-colesterol de 207,7±42.9mg/dl, HDL-colesterol 65,2±20,4mg/dl. La Apo-A 143,1±48mg/dl, Apo-B 114±23,8mg/dl, lipoproteína (a) 30,1±28,6mg/dl. Estudio genético LDLR en 16 niños, hallazgos: 6 mutaciones diversas y 3 delecciones, un caso negativo( LipoCHIP saliva), y 6 pendientes. Estudios grosor del complejo íntima-media, carotideo en 14 ( 7niños y 7 niñas) media 0,6±0,1mm > percentil 75. Hasta el momento han recibido tratamiento farmacológico 5 niños ( 4 con estudio genético positivo) . El colesterol total y LDL-C descendió con dieta de forma significativa, pero sin mantenerse; el descenso fue aún mayor y continuo al asociar tratamiento farmacológico. Conclusiones: El tratamiento farmacológico resultó ser más efectivo que el tratamiento dietético aislado. El grosor íntima media se encuentra aumentado en comparación con datos publicados de niños sanos. La confirmación genética afianza el diagnostico de gravedad y la patología que estos niños presentan desde la gestación. Los estudios ecográficos ponen en alerta del riesgo que corren los niños con dislipemias severas.
MP1d2-002 MODIFICACIONES DEL TEJIDO ADIPOSO SUBCUTÁNEO Y VISCERAL Y DE LA SECRECIÓN DE ADIPOQUINAS EN PACIENTES OBESOS TRAS PÉRDIDA PONDERAL: EFECTO DE LA EDAD Y DESARROLLO G.A. Martos Moreno (1), F.J. Caballero Mora (1), V. Barrios Sabador (2), S. Sirvent Cerdá (3), G. Martínez Díaz-Guerra (4), F. G. Hawkins Carranza (4), J. Argente Oliver (2) (1) Servicio de Endocrinología, Hospital Infantil Universitario Niño Jesús, Instituto de Investigación La Princesa, Madrid; (2) Servicio de Endocrinología, Hospital Infantil Universitario Niño Jesús, Instituto de Investigación La Princesa, Departamento de Pediatría, Universidad Autónoma de Madrid, CIBER Fisiopatología de la Obesidad y Nutrición, Instituto de Salud Carlos III, Madrid; (3) Servicio de Radiodiagnóstico, Hospital Infantil Universitario Niño Jesús, Madrid; (4) Servicio de Endocrinología, Hospital Universitario 12 de Octubre, Departamento de Medicina, Universidad Complutense de Madrid Introducción: Las condiciones fisiopatológicas de los niños obesos varían según su edad y sexo. Los efectos de la reducción ponderal sobre la distribución anatómica del tejido adiposo (TA) y su funcionalidad son insuficientemente conocidos. Objetivos: Describir las modificaciones de la cantidad, distribución y secreción endocrina del TA en niños obesos tras reducción ponderal. Pacientes y métodos: Estudio prospectivo de 32 pacientes (16/sexo) con IMC >+2DE (3,88±1,13), edad: 12,33±2,75 años (6,16-17,25), 8 prepúberes (OB-Pre) y 24 púberes (OB-Pub) (11/13-niñas/niños) al diagnóstico y tras reducción de su IMC >1,5DE. Se determinaron los niveles plasmáticos de leptina y adiponectina total (RIA) y de receptor soluble de leptina (sObR) y HMW-adiponectina (alto peso molecular, ELISA). La distribución corporal e intra-abdominal del TA se estudió mediante DEXA (Hologic-QDR4500W) y resonancia magnética (1,5T-Phillips-Achieva), respectivamente. Resultados: Todos los pacientes disminuyeron su TA subcutáneo (TA-SQ) y visceral (TA-VIS) (p<0,001). La reducción del TA-SQ fue homogénea en OB-Pub, pero no significativa en los miembros inferiores en OB-Pre. La reducción de TA-VIS fue superior a la de TA-SQ en OB-Pre, mientras en los OB-Pub el ratio TA-VIS/TA-SQ no se modificó. Los niveles de adiponectina HMW y total aumentaron en OB-Pre (sólo adiponectina total en OB-Pub); reduciéndose los de leptina sólo en OB-Pub (Tabla). Conclusiones: 1) La reducción de tejido adiposo tras pérdida ponderal en niños obesos no es homogénea y se ve influida por la edad y el desarrollo puberal. 2) La pérdida de peso en edades tempranas determina modificaciones óptimas de la distribución de grasa corporal y del patrón de adipoquinas circulantes.
MP1d2-003 SEDENTARISMO, ESTADO INFLAMATORIO Y DISTRIBUCIÓN GRASA EN LA POBLACIÓN INFANTO-JUVENIL (1) M.G. Bueno Lozano, (2) E. González Gil, (3) J. Olza Meneses, (3) C. Aguilera García, (1) O. Bueno Lozano, (1) J.M. Garagorri Otero, (3) A. Gil Hernández, (2) L.A. Moreno Aznar (1) Endocrinología Pediátrica, HCU Lozano Blesa, Zaragoza, (2) Grupo GENUD Universidad de Zaragoza, Zaragoza, (3) Departamento de Bioquimica y Biología Molecular, Universidad de Granada Introducción: En el adulto obeso, el estado proinflamatorio está relacionado con posibles complicaciones metabólicas y predisposición a enfermedad cardiovascular. Todas estas situaciones de riesgo, pueden tener su origen en la infancia y estar relacionadas con estilos de vida que no favorecen la realización de ejercicio físico de forma regular. Objetivo: Estudiar la relación existente entre marcadores inflamatorios, distribución de grasa corporal y sedentarismo en un grupo de niños y adolescentes. Población estudiada: 121 pacientes (56 varones y 65 mujeres) de edades comprendidas entre 5,1 y 15,3 años. De acuerdo a las tablas de Cole et al. fueron clasificados en: 49 normopeso, 24 sobrepeso y 48 obesos. Entre las variables estudiadas se consideraron: IMC (Kg/m2), tensión arterial, glucemia en ayunas, insulinemia, colesterol, triglicéridos, HDL, LDL, proteína C reactiva (CRP), plasminógeno activador inhibidor 1 total (tPAI1), plasminógeno activador inhibidor (PAI), leptina, interleukina 6 (IL6), resistina y HGF, entre otros. Se calculó el índice HOMA como indicador de resistencia a la insulina. La distribución grasa fue cuantificada mediante DXA (Dual Energy X-Ray Absorptiometry. Para valorar las conductas sedentarias se registraron, mediante cuestionario, las horas al día dedicadas a diferentes actividades de tecnología (televisión, ordenador, consola y móvil). Resultados: El grupo de niños obesos ha presentado cifras de CRP, aPAI y tPA1 significativamente superiores a los niños con normopeso y con sobrepeso (p< 0,05). El contenido de grasa a nivel abdominal obtenido por DXA se ha correlacionado significativamente con HOMA y con los distintos marcadores inflamatorios: resistina, lL-6, CRP, aPAI, tPAI y HGF (p<0,001). Los niveles plasmáticos de los marcadores: aPAI y tPAI, estaban significativamente relacionados con las horas dedicadas a los comportamientos sedentarios, independientemente del IMC (p<0,001). Conclusiones: Estos resultados sugieren que el estado inflamatorio se asocia a la obesidad infantojuvenil y a su distribución central. Además, los comportamientos sedentarios, independientemente del IMC, están relacionados con dichos marcadores de inflamación, todos ellos considerados indicadores de riesgo coronario. De ser así, promover iniciativas dirigidas a evitar hábitos sedentarios en el niño, serían de gran importancia para evitar enfermedad cardiovascular futura.
MP1d2-004 OBESIDAD MÓRBIDA DEL ADOLESCENTE. EXPERIENCIA Y RESULTADOS A MEDIO PLAZO (18-24 MESES) CON EL BALÓN INTRAGÁSTRICO D. Yeste Fernández (1), G. Guillén (2), C. Marhuenda (2), M. Gussinyé (1), V. Martínez-Ibañez (2), A. Carrascosa (1) (1) S. Endocrinología Pediátrica, Hospital Universitario Vall d´Hebron, Barcelona, (2) S. Cirugía Pediátrica. Hospital Universitario Vall d´Hebron, Barcelona INTRODUCCIÓN: El balón intragástrico (BIG) es un método poco invasivo y reversible y puede ser una herramienta útil para reforzar la terapia conductual y el cambio de estilo de vida de los pacientes adolescentes con obesidad mórbida antes de decidir la cirugía bariátrica. OBJETIVOS: Evaluación de los resultados obtenidos a los 18-24 meses del explante del BIG en un grupo de adolescentes con obesidad mórbida. PACIENTES Y METODOS: Se seleccionaron 8 candidatos con edades entre los 13.9 y los 17.9 años, 5 mujeres (M) y 3 varones (V). Todos los pacientes tenían un IMC superior o muy próximo a 40, sufrían una o más comorbilidades graves y cumplían los criterios de selección internacionalmente aceptados. Los BIG (B.I.B. Bioenterics®) se colocaron por vía endoscópica, bajo anestesia general y se mantuvieron durante 6 meses, con una pauta dietética estricta. RESULTADOS: En las Tablas 1 y 2 se muestran los datos clínicos de los pacientes y la evolución del peso, IMC e IMC-zs en el momento del explante del BIG (6 meses) y a los 18-24 meses. Cuatro pacientes mantuvieron en remisión la pérdida ponderal conseguida tras el explante del BIG (pérdida de -11.0 ± 3.7 de IMC). Los restantes recuperaron o incrementaron ligeramente el peso previo al implante del BIG. CONCLUSIONES: En nuestra experiencia el BIG es una metodología segura y con escasas complicaciones. El 50% de la pacientes sujetos a BIG presentan una pérdida significativa y mantenida de peso a los 18-24 meses de su explante.
MP1d2-005 TIROIDECTOMÍA EN PEDIATRÍA: CAUSAS Y COMPLICACIONES L. Gutiérrez Pascual, M.A. Molina, M. Cuesta Rodríguez, B. Huete Hernani, I. Rabanal, I. Gónzalez Casado Hospital la Paz, Madrid Introducción: la extirpación total o parcial de la glándula tiroides es una práctica poco frecuente en pediatría y tiene mayor tasa de complicaciones que en pacientes adultos. Material y métodos: revisión de indicaciones y complicaciones de las tiroidectomías realizadas en nuestro hospital infantil desde 1996 hasta 2011. Resultados: se han realizado 20 intervenciones en dicho periodo, una hemitiroidectomía y 17 tiroidectomías totales (2 de ellas en dos tiempos), siendo los pacientes 13 mujeres y 5 varones. La media de edad es de 7.8 años, con 8 casos < 5 años, 5 casos de 5-10 años y 5 casos de 10-16 años. Desde 1996 hasta 2002 las realizaron cirujanos pediátricos (un total de 3) y posteriormente otorrinolaringólogos infantiles. Las causas comprenden: tiroidectomía profiláctica en portadores de mutación del gen RET (66.6 %), nódulo tiroideo con sospecha de malignidad (4 casos), enfermedad de Graves (1 caso) y bocio multinodular (1 caso). Se produjeron complicaciones en cuatro pacientes, con hipoparatiroidismo e hipocalcemia secundaria (20 %), siendo dos de ellos transitorios (tiempo máximo de recuperación de 5 meses), y otros dos, en los que se realizó tiroidectomía en dos tiempos, continúan en tratamiento tras 3 y 9 meses post-intervención. Uno estos dos casos presentó además parálisis de cuerdas vocales bilateral. Discusión: La tasa de complicaciones es similar a la de otras series, encontrando complicaciones generales en un 5 %, y complicaciones endocrinas en un 20 %, con hipocalcemia permanente entre un 5 y un 10 %, siendo la tiroidectomía en dos tiempos un posible factor de riesgo. Las complicaciones de este tipo de cirugía son más frecuentes en la edad pediátrica. Probablemente se deba a las dificultades anatómicas y a la baja frecuencia con la que este procedimiento se realiza en niños. Es importante que la intervención se lleve a cabo por cirujanos especializados, ya que las tasas de complicaciones descritas son superiores cuando esto no ocurre.
MP1d2-006 ESTUDIO DESCRIPTIVO DE LOS PACIENTES DIAGNOSTICADOS DE HIPOTIROIDISMO CONGÉNITO EN EL CRIBADO NEONATAL QUE PRESENTAN UN COEFICIENTE INTELECTUAL INFERIOR A 2 SDS J. I. Perales Martínez (1), B. Puga González (2), E. Mayayo Dehesa (3), A. De Arriba Muñoz (2), J. I. Labarta Aizpún (3), Á. Ferrández Longás (2) INTRODUCCIÓN Y OBJETIVO: El objetivo del programa de cribado neonatal del hipotiroidismo congénito (HC) es evitar el retraso mental; dicho objetivo se alcanza en la mayoría de los casos pero no en la totalidad. Nuestro trabajo pretende describir las características personales y familiares de los niños que sufren retraso. PACIENTES Y MÉTODO: Desde la implantación en nuestra Comunidad del programa de detección precoz del HC se han diagnosticado 92 casos en la Unidad de Endocrinología Pediátrica (Abril1989-Febrero2011).Estudio descriptivo retrospectivo de los siguientes parámetros en aquéllos pacientes que presentaban en el último control un coeficiente intelectual (CI) <2 SDS. 1). Datos al diagnóstico (TSH screening, dosis y edad inicio tratamiento), 2) Evolución de TSH, FT₄ y CI (Test de Brunet-Lezine, Mc McCarthy y Weschler) 3) Comorbilidad personal, 4) Antecedentes familiares y 5) Aspectos socioeconómicos (Escala de Graffar). RESULTADOS: Los resultados aparecen de forma resumida en la Tabla 1. Diez pacientes presentaron un CI <2SDS (10,8%). El retraso se observó tanto en HC permanente (70%) como transitorio (30%). La edad de inicio de tratamiento osciló entre 4 y 22 días (13,1±4,86). La dosis inicial de L-Tiroxina fue de 8,01±0,8 μg/Kg/día en pacientes diagnosticados antes de 1986 y de 13,85±1,42 μg/Kg/día a partir de esa fecha, salvo un paciente que resultó un HC transitorio. El nivel de FT₄ se normalizó en la 2ª semana en todos los pacientes. En 5 casos (50%) pudieron estar involucrados factores prenatales; 3 retraso de la maduración ósea y 2 hipotiroidismo materno mal controlado durante el embarazo. Otros factores, como la fuerte distocia social en una familia o la presencia de comorbilidades en 3 pacientes (TDAH, depresión y sordera), podrían haber influido en el retraso intelectual. COMENTARIO: Los resultados obtenidos obligan a seguir optimizando la calidad del programa de cribado neonatal del hipotiroidismo, tanto en la etapa prenatal como postnatal, en aras de que el desarrollo intelectual sea porcentualmente similar al de la población general.
MP1d2-007 MUTACIONES EN EL GEN IGSF1 EN NIÑOS CON HIPOTIROIDISMO CONGÉNITO CENTRAL Y MACROORQUIDISMO A. Escudero López (1), R. Barrio (1), D. Gorbenko (2), E. Vallespín (3), J. Nevado (3), P. Lapunzina (3), A. Hokken-Koelega (2), J. C. Moreno (4) (1) Endocrinología pediátrica, Hospital Universitario Ramón y Cajal, Madrid; (2) Pediatric Endocrinology, Sophia Children’s Hospital, Erasmus University Medical Center, Rotterdam, Netherlands; (3) Laboratorio de Genómica Estructural, Instituto de Genética Médica y Molecular (INGEMM), Hostpital Universitario La Paz, Madrid; (4) Laboratorio de Tiroides, Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Madrid. El hipotiroidismo congénito central (HCC) está causado por la deficiencia de TSH. Los genes implicados en esta patología son TSHB y TRHR. Las activinas-inhibinas forman un complejo sistema de factores endo- y autocrinos con función importante en la hipófisis y las gónadas, sin embargo, hasta ahora no se han descrito defectos de estos factores con alteraciones en el eje tiroideo. IGSF1 se ha propuesto como un receptor de membrana de inhibina-B. Objetivo: Investigar las causas moleculares del HCC-Macroorquidismo usando técnicas de secuenciación masiva e Hibridación Genómica Comparada (arrays-CGH). Pacientes y métodos: Se estudiaron 3 pacientes que presentaban HCC-macroorquidismo con herencia ligada al cromosoma X. Tras un estudio hormonal y clínico, se realizó secuenciación masiva del cromosoma X y array-CGH. Resultados: El HCC fue diagnosticado por cribado neonatal basado en T4 o hipotiroidismo clínico (TSHs:1.4-3.9 mU/L; T4L:7.2-8 pmol/L). El test de TRH evidenció una disminución en la respuesta de TSH indicando un hipotiroidismo hipofisario. A los 3-6 años de edad el tamaño testicular estaba por encima de lo normal (3-5 ml Prader). El test de GnRH mostró estimulación de la FSH y de la LH, pero niveles de testosterona indetectables. La pubertad comenzó a los 12.5 años de edad con un volumen testicular inicial de 8 ml (N:2), alcanzando un volumen al final de la pubertad de unos 35-40 ml (N:20-25). La inhibina B (425-500 ng/L; N:200-400) y hormona antimuleriana (19-48 µg/L;N:5-9) estaban elevadas lo que sugiere un incremento de células de Sertoli. En el paciente 1, un array CGH mostró una deleción de 200 kb que abarcaba el gen IGSF1 completo. En pacientes 2 y 3 la secuenciación masiva del cromosoma X reveló una mutación puntual en IGSF1 (p.C942R) que cosegregaba con el fenotipo en la familia. Conclusiones: IGSF1 es un nuevo gen candidato para el hipotiroidismo central. Proponemos que defectos en IGSF1 pueden alterar las funciones de la hipófisis y del testículo debido al desequilibrio entre inhibina-B/activina-A que lleva a una alteración en vías de señalización de células tirotropas, gonadotropas y/o de Sertoli.
MP1d2-008 TEST DE ACTH: ¿ES DE UTILIDAD EN EL DIAGNÓSTICO DE LAS FORMAS DE INICIO TARDÍO DE HIPERPLASIA ADRENAL CONGÉNITA? D. Bareño Campos, L. Losada Burbano; D. Yeste Fernández; M. Gussinye Canadell; A. Carrascosa Lezcano; M.Clemente León Hospital Vall d'Hebron, Área Materno-Infantil, Barcelona Introducción: La pubarquia prematura es la manifestación más habitual de la hiperplasia adrenal congénita no clásica (HSC-NC), la aceleración de la velocidad de crecimiento y/o de la edad ósea en edad prepuberal, el hirsutismo y la hipertrofia de clítoris pueden presentarse clínicamente. El test de estímulo adrenal con ACTH es la metodología recomendada para diagnosticar las formas de HSC de presentación tardía. Objetivo: Determinar la especificidad y la sensibilidad de las concentraciones plasmáticas basales de predictores de HSC-NC (17-OH-Progesterona (17-OHPG), androstendiona, DHEA-S y testosterona. Pacientes y métodos: Estudio retrospectivo de cohorte en el que se incluyeron los pacientes (n=280) con indicación de test de ACTH por sospecha de HSC-NC. Se considero criterio diagnóstico de HSC-NC la respuesta post-ACTH (60 min.) de 17-OHPG³10 ng/ml con confirmación de genética molecular. Análisis descriptivo univariante y curvas de ROC para determinar la sensibilidad y la especificidad de cada uno de los parámetros evaluados. Resultados: Las tablas muestran los motivos que indicaron la práctica del test de ACTH, sexo, edad media a la que se efectuaron y el número y porcentaje de resultados positivos. Conclusiones: El 10,7% de los pacientes fueron diagnosticados de HS-NC y confirmados por genética molecular. Las concentraciones plasmáticas basales de testosterona, androstendiona y DHEA-S no fueron de utilidad para identificar los pacientes con HSC-NC. Las concentraciones plasmáticas basales de 17-OHPG ³ 2ng/ml tienen un 100% de sensibilidad y un 95% de especificidad para el diagnóstico de HSC-NC, haciendo innecesaria la práctica de un test de ACTH.
MP2d3-009 RESISTENCIA A IGF-I DEBIDA A HAPLOINSUFICIENCIA DEL RECEPTOR DE IGF-I Y RESPUESTA AL TRATAMIENTO CON rhGH E. Gallego Gómez (1), J. Sánchez del Pozo (1), J. Cruz Rojo (1), A. Gómez Núñez (2), R. Gracia Bouthelier (3), K. Heath (2, 4) A. Campos Barros (2, 4) (1) Hospital Univ. 12 de Octubre, Servicio de Endocrinología Pediátrica, Madrid; (2) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hospital Universitario La Paz, Madrid; (3) Servicio de Endocrinología Pediátrica, Hospital Universitario La Paz, Madrid; (4) Centro de Investigación Biomédica en Enfermedades Raras (CIBERER, U753), Instituto Carlos III, Madrid. Introducción: El síndrome de resistencia a IGF-I (SR-IGF1) se caracteriza por retraso del crecimiento intrauterino y postnatal con niveles normales o supranormales de IGF-I, microcefalia acompañada de rasgos dismórficos y déficit intelectual variable. SR-IGF1 está causado por anomalías genómicas o mutaciones que afectan al gen IGF1R (15q26.3). Caso clínico: Niña nacida a término mediante cesárea por CIR simétrico con PRN: 1.850 g (-3,84DE), talla 42 cm (-4.71DE) y PC 30 cm (-3.21DE); talla madre 159,5 cm; (-0,75DE); talla padre: 172,3 cm; -0,78DE). Con 1,7 años presentó déficit del crecimiento ponderoestatural, peso: 7,2 Kg. (-3.38DE), talla: 70,5 cm (-4,17DE), PC: 42 cm (-4,54DE), así como desproporción craneofacial leve, clinodactilia 5º dedo bilateral, CI de 69%, retraso de la EO de -1DE e IGF-I: 118,0 ng/ml (-0,69DE). A los 4,5 años (talla -4,24DE; peso: -1,86DE), EO 3 años, BMI:+0,42DE e IGF-I:279 (+1,82DE), inició tratamiento con rhGH (0,035 mg/kg/d) por indicación CIR sin estirón recuperador, obteniendo una curva de crecimiento ascendente con elevación progresiva de los valores de IGF-I e IGFBP3. Estudios genéticos: cariotipo; análisis molecular de IGF1R mediante HRM y secuenciación y MLPA (deleciones/duplicaciones). Resultados: Deleción completa de novo de IGF1R en heterocigosis en índice. Seguimiento: Tras tres años de tratamiento, presentó talla de 114,4 cm (-2.5 DE), velocidad de crecimiento 5,68 cm/año, con IGF-I muy elevada: 1.066 ng/ml (+4,74DE) e IGFBP3: 8.09 mg/ml (+2.71DE). Tras suspensión del tratamiento debido al marcado incremento de IGF-I, los 6 primeros meses mantuvo velocidad de crecimiento de 7,2 cm/año con descenso marcado de IGF-I (367 ng/ml; +2DE) e IGFBP3 (643 mg/ml; +1.8DE), disminuyendo progresivamente a 3,09 cm/año durante el 2º semestre sin tratamiento y manteniéndose en los últimos 6 meses a 3,38 cm/año, con talla actual: 121,5 cm (-2,46DE) e incremento llamativo de BMI:22.73 y de grasa troncular y miembros superiores. Conclusiones: La haploinsuficiencia de IGF1R por deleción completa de IGF1R en heterocigosis causa un fenotipo característico de SR-IGF1. El tratamiento con dosis bajas de rhGH normaliza la velocidad de crecimiento elevando notablemente la IGF-I circulante. Es necesario ponderar los posibles riesgos de dicho incremento con los beneficios del tratamiento en estos pacientes.
MP2d3-010 ¿EXISTEN PARÁMETROS PREDICTIVOS DE LA RESPUESTA AL TRATAMIENTO CON HORMONA DE CRECIMIENTO EN PACIENTES CON RETRASO DEL CRECIMIENTO INTRAUTERINO? A. Rodriguez Estevez (1), C. Fernandez Ramos (2), L. Martinez-Indart (3), I. Rios Orbañanos (1), B. Pacho del Castaño (1), I. Rica Etxebarria (1), A. Vela de Sojo (1), G. Grau Bolado (1), F.J. Nuñez Rodríguez (2), I. Diez López (4), A. Sarasua Miranda (4), E. Artola Aizalde (5), Cancela Muñiz (5), E. Blarduni Cardón(6), A. Eguireun Rodríguez (7) (1) Endocrinología Pediátrica, Hospital Universitario de Cruces; (2) Hospital Universitario de Basurto; (3) Unidad de Epidemiología Clínica Hospital Universitario de Cruces; (4) Hospital Universitario de Alava; (5) Hospital Universitario de Donosti; (6) Hospital de Zumárraga, (7) Hospital de Mendaro. Objetivos: 1-Describir la población con Retraso del Crecimiento Intrauterino (RCIU) en tratamiento con Hormona de Crecimiento (GH) en nuestra Comunidad. 2-Valoración de la respuesta al tratamiento en función de distintas variables. Pacientes y Metodología: Recogida de datos retrospectiva de los pacientes RCIU en tratamiento con GH. Fuente: protocolos de GH del Comité. Se excluyen niños afectos de Síndrome de Silver-Russell. Descripción de variables categóricas (%) y continuas (media±DE) Comparación de diferentes variables y VC (prueba no paramétrica de Kruskal-Wallis). Resultados: 87 pacientes (43V, 44M) RCIU. Longitud al nacimiento (Ln) 42.9±4.3 cm (-2.8±1.2) y Peso (Pn) 2067,9±638 gr (-2.1±0.7DE). Ninguno presentaba fenotipo peculiar y 5 referían retraso psicomotor. 31% pretérminos (<37 semanas de gestación). Talla diana en niñas 154.7±4.7 cm (-1.5±0.8DE) y en niños 170.8±4.5 cm (-0.9±0.8DE). 16 % presentaban antecedentes familiares de retraso puberal. Evolución en DE de talla (T), peso (P), velocidad de crecimiento (VC), y edad ósea (EO por Greulich-Pyle) se muestran en la tabla 1. Establecimos 3 grupos en función de la VC en el primer año de tratamiento :<1DE: n=17; 1.1-2 DE: n=14; >2.1 DE: n=56. Al comparar los 3 grupos no encontramos ninguna asociación con la talla paterna y materna, Ln y Pn, edad gestacional (pretérmino/a término), parámetros iniciales (talla, EC y EO, VC) ni con la dosis de GH, similar en los 3 grupos (34.5/35.1/33.6 mcg/Kg/d). El grupo de niños con AF de talla baja (28%) tenía una talla inferior al inicio del tratamiento (-3.3±0.6 versus -3.0±0.5) -p.040- que el grupo con TF normal. Además la VC del primer año fue menor en este grupo aunque no alcanzo diferencias significativas (2.0±1.7 versus 2.7± 1.4DE) -p .081- . Conclusiones: 1-La respuesta al tratamiento ha sido favorable con una ganancia de talla de 0.5 y 1.2 DE al año y 4 años de tratamiento. 2-No hemos encontrado parámetros predictivos de mejor respuesta al tratamiento con GH.
MP2d3-011 EXPERIENCIA EN EL TRATAMIENTO CON HORMONA DE CRECIMIENTO EN PACIENTES CON SÍNDROME DE PRADER-WILLI EN UN CENTRO DE REFERENCIA I. Iglesias Rodríguez (1), R. Corripio Collado (1), J. Pérez Sánchez (1), E. Gabau Vila (2) (1) Unidad Endocrinología Pediátrica, (2) Unidad Genética Clínica, Hospital de Sabadell, Corporació Sanitària Parc Taulí, Institut Universitari Parc Taulí, Universitat Autònoma de Barcelona. Introducción Desde el año 2000 está aprobado el tratamiento con hormona de crecimiento (GH) en el Síndrome de Prader-Willi (SPW). Dicho tratamiento mejora la composición corporal, crecimiento, fuerza muscular y función pulmonar. Nuestro hospital es centro de referencia de SPW desde 1997. Objetivo Analizar el efecto del tratamiento con GH en el crecimiento, talla final e índice de masa corporal (IMC) en los pacientes afectos de SPW de nuestro centro, así como sus posibles efectos adversos. Material y métodos Estudio descriptivo retrospectivo de los pacientes con SPW de nuestro centro, nacidos entre 1980- 2011. Se analizan talla y peso anualmente, desde su llegada al centro hasta la edad adulta. Con los datos antropométricos se calculan velocidad de crecimiento e IMC. Se comparó los pacientes afectos de SPW tratados con GH (dosis 0,03 mg/kg/día) con los no tratados. Se recogió la aparición o empeoramiento de escoliosis, hipotiroidismo, diabetes mellitus, apneas obstructivas y episodios fatales. Resultados Se han analizado los datos de 25 pacientes (12 varones) de edad media 14,8 años (rango 1,28-35,2) de los cuales 15 han recibido GH. La talla final media de los 12 pacientes que ya la han alcanzado es de -1,58±1,06 DE en el grupo de tratados (n=6) y de -2,88±0,86 DE en los no tratados (diferencia -1,3; IC95%: -2,54 a -0,05). El IMC medio de los pacientes tratados con GH a los 18 años fue de 26,1±4,2 Kg/m2 y de los no tratados de 38,3±10,7 kg/m2 (diferencia 12,1; IC 95%: 2,3-21,99). En el primer año de tratamiento se objetivó un incremento de la talla en los paciente tratados (n=15) en 0,65 DE (10,5 cm). Además 7 presentaban escoliosis pretratamiento y en 1 caso empeoró. De los 10 no tratados, 8 presentaban escoliosis. En ninguno de los pacientes que recibieron GH se objetivó aparición de hipotiroidismo, diabetes mellitus 2, apneas obstructivas ni episodios fatales. Uno no tratado presentó diabetes tipo 2. Conclusiones Hemos objetivado una mejoría en la talla final e IMC sin complicaciones. La evidencia de un efecto beneficioso de GH en la mayoría de niños con SPW es tan fuerte que debería ser considerado en todos ellos, realizando un seguimiento multidisciplinar para minimizar los posibles efectos adversos.
MP2d3-012 PATRONES DE VARIACIÓN GENÉTICA EN LA REGIÓN PRADER-WILLI, ASOCIADOS A OBESIDAD INFANTIL SEVERA P. Méndez Pérez (1), R. Rodríguez-López (2), J. Sáenz (2), J.M. Carbonell (2), A. Margallo Balsera (2), M. González-Carpio Serrano (2), M. García de Cáceres (2), T. Herrera Moreno (2) , P. Sánchez Giralt (3), E. Galán Gómez (1), M. Núñez Estévez (1) (1) Departamento de Pediatría, Hospital Materno-Infantil Infanta Cristina, Badajoz; (2) Hospital Infanta Cristina, Unidad de Genética, Badajoz; (3) Hospital Infanta Cristina, Servicio de Endocrinología, Badajoz INTRODUCCIÓN: La susceptibilidad heredada a padecer obesidad severa y desde la infancia, se asocia progresivamente a la variabilidad interindividual de genes y regiones relacionadas con el sobrepeso. Los análisis de ligamiento a partir de los datos del proyecto HapMap se tornan esenciales para identificar las alteraciones funcionales, agentes causales de riesgos congénitos. Analizamos la región crítica del Síndrome de Prader-Willi (15q11-q13), base molecular de las alteraciones neuroendocrinas que generan un sobrepeso grave en estos pacientes. Estudios recientes de asociación en el genoma completo (GWAS) señalan la capacidad deletérea de la variación de número de copias (CNVs) en esta región, por modificar su compleja regulación. PACIENTES Y MÉTODOS: Usando la plataforma Sequenom, hemos genotipado 49 SNPs contiguos en la región del Síndrome de Prader-Willi, en 218 niños afectados de obesidad severa y que pertenecen a familias que refieren antecedentes de alto riesgo, así como en 198 controles. RESULTADOS: Las diferencias en la distribución de los genotipos (p = 0,00234), los patrones de desequilibrio de ligamiento de la región y de la diversidad de sus haplotipos (p = 0,0099) identificaron perfiles comunes de susceptibilidad genética a la ganancia de peso desde la infancia. CONCLUSIONES: La selección correcta de pacientes con sobrepeso de inicio temprano y familiar, maximiza el poder estadístico de los estudios de asociación y reduce el tamaño de la muestra precisa. Se ha atribuido una predisposición a desarrollar obesidad a la variación estructural (duplicaciones o deleciones polimórficas) de tres segmentos (de 2 a 23 Kb) localizados en esta región, que se encuentran y se superponen exactamente con el bloque 1 de desequilibrio en el que encontramos los SNPs de riesgo en nuestra serie. En base a estas CNVs se ha descrito la modificación de la expresión de ciertos genes de la región, mediante la interrupción de las secuencias codificantes, perturbando su regulación o alterando la dosis génica. Los análisis de CNVs pueden facilitar la comprensión de las bases genéticas de la obesidad y sugieren que la integración de estos datos con los de variantes tipo SNP, podrían aumentar la eficiencia de la investigación en la heredabilidad del sobrepeso grave. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites