
| Rev Esp Endocrinol Pediatr 2012;3 Suppl(1):156-175 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2012.Apr.117 |
| Diabetes |
| Sent for review: 17 Apr. 2012 | Accepted: 17 Apr. 2012 | Published: 30 Apr. 2012 |
| Congreso SEEP |
| Correspondence:Congreso SEEP |
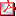 Tabla - P1d2d3-061 Tabla - P1d2d3-061 |
| Tabla - P1d2d3-070 |
| Tabla - P1d2d3-074 |
| Tabla - P1d2d3-075 |
| Tabla - P1d2d3-078 |
| Tabla 1 - P1d2d3-057 |
| Tabla 1 - P1d2d3-065 |
| Tabla 1 - P1d2d3-069 |
| Tabla 1 - P1d2d3-082 |
| Tabla 2 - P1d2d3-057 |
| Tabla 2 - P1d2d3-065 |
| Tabla 3 - P1d2d3-065 |
| Tabla 4 - P1d2d3-065 |
| Tabla 5 - P1d2d3-065 |
 Figura - P1d2d3-080 Figura - P1d2d3-080 |
|
|
P1d2d3-054 ALTERACIONES CUALITATIVAS Y CUANTITATIVAS EN LA COMPOSICION DE LOS ISLOTES PANCREATICOS DE UNA PACIENTE CON HIPOGLUCEMIA NEONATAL REBELDE A TRATAMIENTO C. Moreno Jiménez, (1), L. Mª Sáez García, (1), P. Ruiz Ocaña, Pablo (2), L. Escobar Luis (3), I. Cozar Castellano, (3), A. M. Lechuga Sancho (1) (1) Departamento Materno-Infantil y Radiología. Facultad de Medicina. Universidad de Cádiz. Cádiz, (2) Unidad de Investigación Hospital Universitario Puerta del Mar. Cádiz, (3) Unidad de gestión clínica de pediatría. Hospital Universitario de Puerta del Mar. Cádiz Introducción: La hipoglucemia neonatal es una entidad clínica de etiología y curso clínico variable. En la actualidad se esta avanzando en el conocimiento de los genes implicados en muchos de estos casos, sin embargo es escasa la literatura en su descripción anatomopatológica. Presentamos el caso de una paciente con hipoglucemia neonatal, resistente a tratamiento a la que no se encontró defecto molecular conocido, y a la que tuvimos la oportunidad de estudiar anatomopatológicamente. Paciente y métodos: Mujer de 28 Semanas de gestación y peso adecuado a su edad gestacional que presenta desde el primer día hipoglucemias sintomáticas con hiperinsulinemia, que precisaba aportes i.v. de glucosa de 36 mg/kg/min. Recibió tratamiento con hidrocortisona (10mg/Kg./día), diazóxido, y somatostatína (hasta 10 mcg/kg./día), asociada a glucagón. A los 28 días, comienza a alternar periodos de hipo e hiperglucemia, por lo que se retira el tratamiento. Exitus a los 73 días por fallo multiorgánico. El páncreas se dividió en cabeza y cola y se fijo e incluyo en parafina. Se cortaron lonchas de 5 micras y se tiñeron con anticuerpos frente a insulina, glucagón, somatostatína y TUNEL para apoptosis. Se visualizaron en microscopio de fluorescencia con cámara, y se cuantificaron al menos 20 campos escogidos al azar, en cada fragmento del páncreas. Resultados: En la cabeza del páncreas se encontraron un mayor número de islotes, de tamaño normal, pero marcaje de insulina irregular. Encontramos un numero cinco veces mayor de células insulina-positivas en la cabeza (p<0,001), mientras que no hubo un mayor numero de células glucagón positivas (p=0,118). Curiosamente, no se encontraron células teñidas con somatostatína en la cabeza, pero si en la cola. Hubo una mayor tasa de células beta apoptoticas (TUNEL positivas), en la cabeza. Conclusiones: El mayor numero total de células beta por área, y el mayor numero de estas en apoptosis, sugiere que el turnover de la célula beta en la cabeza del páncreas de la paciente esta muy acelerado. No podemos descartar la posibilidad de que esta muerte se deba en parte a los tratamientos
P1d2d3-055 CONVULSIÓN NEONATAL SECUNDARIA A HIPOGLUCEMIA EN EL CONTEXTO DE HIPERINSULINISMO CONGÉNITO J.L. Castellano Pérez, M. López García, M. C. Ontoria Betancort, Mª I.García Camiñas, J. Ramón Castro Conde, J. P. González Díaz Complejo Hospitalario Universitario de Canarias. INTRODUCCIÓN: El hiperinsulinismo congénito es la causa más frecuente de hipoglucemia persistente en el lactante. Es resultado de una secreción insulínica anómala, expresión de varias alteraciones genéticas, siendo el estudio compatible en un 50% de los casos. Se distinguen dos tipos en función de la afectación pancreática, focal o difusa, lo cual es importante de cara al tratamiento y pronóstico. CASO CLÍNICO: Neonato mujer con estado convulsivo clónico multifocal a los dos días de vida secundario a hipoglucemia capilar de 16 mg/dl. Se detecta episodios persistentes de hipoglucemias asintomáticas más allá de la primera semana de vida con aportes endovenosos de glucosa de hasta 15 mg/kg/minuto. EF: Normal. AP: RNT (39 semanas), AEG (3380 gramos) sin incidencias perinatales ni otras neonatales. Exámenes complementarios: Analítica: glucosa 32 mg/dl, insulina basal 6.9 mcU/ml, péptido C basal 2.85 ng/ml, cortisol basal 15.10 mcgr/dL, GH basal 23.90 ng/mL, amonio 78 mcgr/dL, ácido pirúvico 97 mcmol/L, ácido láctico 4.1 mcmol/L, transaminasas, cetonemia y cuerpos reductores en orina negativos. Test de glucagón: elevación de la glucosa 35 mg/dL respecto a la basal. Aminoácidos en sangre y orina: normales. Estudio molecular para genes KCNJ11 y ABCC8: negativos. Ecografía transfontanelar y de abdomen: normales. iRM craneal y abdominal: normales. PET/TC 18F-fluoro-L-Dopa: imagen nodular hipercaptante en cabeza pancreática limítrofe para un hiperinsulinismo focal, no pudiendo descartarse difuso. Evolución: ante el diagnóstico de hiperinsulinismo se inicia alimentación enteral suplementada con dextrinomaltosa por sonda nasogástrica a débito contínuo, diaxózido e hidroclorotiazida. Glucemia monitorizada de manera contínua con sensor subcutáneo, precisando de forma puntual glucagón intramuscular. Tolera la suspensión de perfusión endovenosa de glucosa al mes de vida. Tres meses después se realiza pancreatectomía parcial (cabeza, proceso uncinado y parte del cuerpo. Anatomía patológica: parénquima pancreático con nesidioblastosis difusa). Tras ello se consigue retirar el tratamiento farmacológico manteniéndose los suplementos de hidratos de carbono de absorción lenta. Desarrollo neurológico adecuado. COMENTARIOS: 1. Es importante filiar la causa de episodios recurrentes de hipoglucemia rebeldes al manejo convencional, dado el alto riesgo de daño neurológico irreversible potencial. 2. Se ha de aplicar un adecuado algoritmo diagnóstico teniendo siempre presente el hiperinsulinismo congénito como posibilidad.
P1d2d3-056 MACROSOMIA E HIPOGLUCEMIA NEONATAL PERSISTENTE SECUNDARIA A NUEVA MUTACIÓN DOMINANTE EN EL GEN ABCC8 F. Hermoso López, G. Sacoto Erazo, JV. Martínez Robles, P. Bahillo Curieses, P. García Gutiérrez, E. Moreno Gómez Hospital Clínico Universitario, Valladolid La macrosomía neonatal e hipoglucemia secundarias a hiperinsulinismo congénito puede ser secundaria a un grupo de enfermedades heterogéneas, por alteraciones genéticas específicas. En el 50% de los casos el defecto estructural se encuentra en el canal de potasio (KATP) de la célula b pancreática, que implica a las subunidades SUR1 y Kir 6.2, a su vez codificadas por los genes ABCC8 y KCNJ11 respectivamente. Se han descrito más de 150 mutaciones en ABCC8 y 25 en KCNJ11, la mayoría recesivas, aunque también se han detectado casos con herencia dominante. La forma dominante presenta unas manifestaciones clínica- hormonales menos graves y la respuesta al tratamiento con diazóxido no suele ser favorable en todos los casos Caso clínico Neonato nacido a las 35 semanas por riesgo de pérdida de bienestar fetal. Embarazo controlado con diagnostico gestacional de intolerancia a la glucosa. Al nacimiento destaca macrosomía fetal (peso: 3.555g (+2.58DE), talla: 51cm (+2.06DE)), rubicundez, facies de luna llena, hipoglucemia severa (18mg/dl) que requiere para su normalización varios bolos de suero glucosado y elevado ritmo minutado de glucosa en las primeras 24 horas de vida (11mg/kg/min) y administración de hidrocortisona durante cuatro días. Se descartan déficits de GH, cortisol y otras alteraciones metabólicas, presenta glucemia de 20mg/dl con insulinemia concomitante de 26uU/ml. La historia familiar reveló diabetes en tres generaciónes previas, miembros macrosomicos al nacimiento y muertes neonatales de causa no esclarecida. El estudio genético de ABCC8 demostró una nueva mutación sin sentido A1537V de herencia dominante. El paciente se manejó con nutrición enteral continua que se pudo interrumpir a las ocho semanas de vida. Conclusiones La macrosomia con hipoglucemia e hiperinsulinemia neonatal persistente obliga al estudio de patología enzimática de célulab. Se realiza diagnostico diferencial con las seis mutaciones más frecuentes para realizar el tratamiento adecuado en corto plazo evitando tratamientos agresivos de por vida como la pancreatectomia y secuelas cerebrales por hipoglucemias repetidas. Las formas dominantes suelen ser más leves y pueden evolucionar con tratamiento sintomático. Se ha sugerido que las mutaciones del canal KATP pueden originar diabetes en etapa adulta por apoptosis de la célula b secundaria a una elevada concentración intracelular de calcio.
P1d2d3-057 HIPERINSULINISMO CONGÉNITO. REVISIÓN DE 16 CASOS. M.C. De Mingo Alemany, N. Pons Fernández, A. Adell Sales, B León de Zayas, S. León Cariñena, F. Moreno Macián Hospital La Fe, Valencia INTRODUCCIÓN: El hiperinsulinismo congénito tiene una incidencia de 1/30000-50000. La causa genética se identifica en un 50% de casos, la disfunción del canal de potasio de la célula beta pancreática es la causa más frecuente. MATERIAL Y MÉTODOS: Estudio descriptivo de pacientes con hiperinsulinismo congénito del Hospital La Fe, desde 1992 hasta 2012. La variables categóricas se describen con % . Las variables cuantitativas continuas normales (Kolmogorov-Smirnov >0.5) con media y desviación típica. RESULTADOS: Muestra: 16 pacientes (6 mujeres, 10 varones) . Edad media al diagnóstico 364.69 días (DS 546.97). Peso al nacimiento media 3517.69 gr (DS 761.42). Cuatro pacientes asocian poliquistosis renal. Una fallo hepático fulminante. Datos al diagnóstico (ver tabla 1). En todos ACTH/cortisol, GH y alanina normal. Cetonuria negativa. Test de glucagón realizado en 6 pacientes. Inicialmente 6 precisan nutrición enteral a débito continuo. Aporte máximo de glucosa intravenosa: media 9.32 mg/Kg/min (DS 7.29). Tratamiento médico (ver tabla 2). En 9 pacientes se suspende el tratamiento, a una media de 5.8 años (DS 5.98). Una paciente fallece al 1.5 años del diagnóstico tras dos pancreatectomías. Otra a los 5 meses del diagnóstico por sepsis fúngica. Efectos secundarios: 16 hipertricosis, 6 retención hídrica, 1 enterocolitis necrotizante. Se realizan 2 PET con L-Dopa fluorada, compatibles con hiperinsulinismo difuso. Se extrae estudio genético a 12 pacientes. 3 mutación en ABCC8 en heterocigosis. 7 estudios negativos para ABBCC8, KCNJ11 y GCK. Dos pacientes asocian retraso psicomotor. DISCUSIÓN: La evolución de nuestros pacientes es similar a la descrita en la literatura. Pensamos que en los casos de fracaso al tratamiento médico sería recomendable realizar PET con L-Dopa-fluorada como exploración de primera línea, dada su utilidad de cara a tomar otras actitudes terapéuticas.
P1d2d3-058 SÍNDROME DE HIPERINSULINISMO CONGÉNITO POR MUTACIÓN DEL RECEPTOR SUR1 M.I. Gálvez Aguilar (1), R. Espino Aguilar (2), L. Acosta Gordillo (3), L. Castaño González (4) (1) Unidad de Endocrinología Pediátrica. UGC Pediatría y Neonatología. Hospital Universitario Ntra. Sra. de Valme. Sevilla; (2) Unidad de Investigación. Endocrinología Pediátrica. Hospital Universitario de Cruces. Vizcaya. INTRODUCCIÓN El hiperinsulinismo (HI) es la causa más frecuente de hipoglucemia persistente durante el primer año de vida. La clínica es precoz y puede llevar a confusión con otras patologías graves del RN. Precisa elevados aportes de glucosa. Las cifras de insulina y péptido C pueden ser normales lo que dificulta el diagnóstico precoz. La tasa de secuelas neurológicas irreversibles es alta. CASO CLÍNICO RN mujer, 1ª gestación de padres no consanguíneos. Madre diabética insulinodependiente. Parto en la semana 37 de gestación. Apgar 9/10. P: 4020 gr (p97), L: 53 cm p90), PC: 34 cm (p50). Desde el nacimiento presentó hipoglucemias severas sintomáticas, ingresó en UCIN y precisó elevados aportes de glucosa (12 mg/kg/min). En estudio protocolizado sólo se evidenció ligera elevación de insulina y péptido C con cifras persistentemente bajas de glucosa. La sospecha de hiperinsulinismo congénito la confirmó el estudio genético que mostró una mutación en el exón 33 del gen ABCC8 (receptor SUR1 pancreático); igualmente se identificó en la madre y abuela materna (ambas con antecedentes de hipoglucemias y diabetes insulinodependiente). Comenzó tratamiento con diazóxido con buena respuesta. RMN mostró atrofia córtico-subcortical, clínicamente destacaba hipotonía generalizada. Con 2.6 años, el desarrollo ponderoestatural y psicomotor son adecuados. Diazóxido en retirada por comenzar con cifras de glucemia ligeramente elevadas. COMENTARIO El HI congénito más común es por alteración en el canal de KATP de la célula betapancreática; 50-60% de casos por mutación del gen SUR1 (ABCC8). La diferenciación histológica determina el pronóstico. Las formas difusas (más graves), con herencia AR y pérdida completa de actividad del gen, son resistentes al tratamiento médico (diazóxido como elección) y pueden precisar pancreatectomía total. Las formas focales (más frecuentes), de presentación esporádica, suelen responder a la medicación y sufrir apoptosis del área afecta con desaparición de la hipoglucemia; si el área apoptótica es muy grande, aparece intolerancia hidrocarbonada o diabetes antes de la adolescencia. Nuestra paciente a pesar de la gravedad y difícil manejo inicial, está evolucionando muy positivamente. Destacar la necesidad de establecer un tratamiento intensivo y protocolizado en estos casos aún en ausencia de confirmación de estudios moleculares.
P1d2d3-059 SÍNDROME DE HIPERINSULINISMO-HIPERAMONIEMIA. ¿EXISTE UNA CORRELACIÓN GENOTIPO-FENOTIPO? A.P. Nso Roca (1), F. Carratalá Marco (1), JM. Donate Legaz (2), A. De la Morena Campillo (1), M. Juste Ruiz (1) (1) Hospital Universitario San Juan de Alicante. Alicante, (2) Hospital Santa Lucía. Cartagena El síndrome de Hiperinsulinismo-hiperamoniemia (HI-HA) es una forma de hiperinsulinismo congénito. Se caracteriza por episodios recurrentes de hipoglucemia sintomática junto con elevación persistente de las cifras de amonio. Se debe a mutaciones en los exones 6, 7, 11 y 12 del gen GLUD1 (cromosoma 10q) que codifica la enzima mitocondrial glutamato deshidrogenasa. Se ha descrito su asociación con epilepsia en casos con mutaciones en los exones 6 y 7 pero la sintomatología clínica es heterogénea. Presentamos un caso de HI-HA asociado a epilepsia e hipocrecimiento. Se trata de una niña, sin antecedentes perinatales de interés, diagnosticada de HI-HA a los 7 meses tras un episodio de hipoglucemia sintomática (29 mg/dl) con insulina 7,9 µUI/ml y amonio 187 µmol/l. La exploración física es normal salvo por la presencia de una mancha café con leche en todo el hemiabdomen derecho. Inicia tratamiento con diazóxido con buena respuesta y el estudio genético confirma una mutación en el exón 11 del gen de la glutamato deshidrogenasa. A los 5 años de vida, desarrolla una epilepsia mioclónica controlada parcialmente con lamotrigina y levetiracetam. A partir de los 5,5 años de edad (talla 105 cm, -1,8 SDS), inicia una disminución de la velocidad de crecimiento con una talla a los 10 años de 122 cm (-3,06 SDS). Se realiza un test de clonidina, con un pico de GH de 5,8 ng/ml por lo que se remite para valoración de tratamiento con hormona de crecimiento. El síndrome HI-HA es una entidad a considerar en el diagnóstico diferencial de los hiperinsulinismos congénitos ya que su detección temprana evita la agresividad en el diagnóstico y en el manejo. Suele presentar buena respuesta inicial al diazóxido pero, evolutivamente, pueden surgir patologías asociadas. Nuestro caso describe la asociación con epilepsia en una paciente sin mutación en los exones 6 o 7. Asocia, además, talla baja patológica siendo, hasta donde sabemos, el segundo caso descrito en la literatura. El seguimiento a largo plazo de estos pacientes y la evaluación de su comorbilidad esclarecerá si existe una correlación entre genotipo y fenotipo.
P1d2d3-060 MUTACIÓN ACTIVADORA DEL GEN DE LA GLUCOQUINASA COMO CAUSA DE HIPERINSULINISMO, A PROPÓSITO DE UN CASO M. Murillo Vallés, J.Bel Comós, J.Herrero Espinet, J. Mª Mengívar Garrido Hospital Universitari Germans Trias i Pujol, Badalona. El hiperinsulinismo congénito (HC) es una entidad poco frecuente (1/35.000-40.000 personas/año) y que presenta gran heterogeneidad clínica y analítica. La disregulación en la secreción de insulina propia de esta entidad puede ser debida a diversas alteraciones en las distintas estructuras que componen la célula beta. La mayoría de casos descritos son por mutaciones en los genes que codifican los componentes del canal de potasio (SUR1 y Kir6.2), pero existen en la literatura casos de HC por mutaciones en otros enzimas como la glutamato deshidrogenasa o la glucoquinasa (GCK). La GCK es considerada el “sensor” de glucosa de la célula beta ya que actúa como regulador de la secreción de insulina. En el caso del HC debido a una alteración en este enzima (hiperactivación), los niveles de glucosa a los que se segrega insulina son menores de lo habitual (al contrario como ocurre en la diabetes MODY tipo 2). Este tipo de HC acostumbra a ser más leve que el debido a otras alteraciones y a presentar una evolución favorable con manejo simplemente farmacológico. Hasta la fecha hay descritas 11 mutaciones activadoras de este gen. Presentamos el caso de una paciente afecta de HC por una mutación activadora del gen de la GCK en el exón 2 (T65I) en heterozigosis. Se trata de una paciente sana sin otros antecedentes relevantes más que macrosomía al nacimiento, que a los 3 años de edad en el contexto de un proceso febril, presenta hipoglucemia hipocetósica persistente (40-50mg/dl) y niveles elevados de insulina. Precisa glucosa parenteral continua para conseguir normoglucemia (6mg/kg/min). Ante la sospecha de hiperinsulinismo se inicia tratamiento con diazóxido (15mg/kg/día) que permite reducir progresivamente la glucosa parenteral hasta suspenderla. Éste se mantiene tras la confirmación genética del tipo de HC. Actualmente, 2 años después del diagnóstico, la paciente continua con diazóxido (6mg/kg/día), sin presentar episodios significativos de hipoglucemias ni efectos secundarios propios de la medicación. El desarrollo pondoestatural y psicomotor es correcto.
P1d2d3-061 IDENTIFICACIÓN DE UNA NUEVA MUTACIÓN EN EL GEN DE LA GLUCOQUINASA EN UNA FAMILIA CON HIPERGLUCEMIA C.M. López López (1), E. Palomo Atance(1), MJ Ballester Herrera(1), E. Martín Campagne(1), P. Giralt Muiña(1), MP López Garrido(2) (1) Hospital General Universitario de Ciudad Real. Ciudad Real; (2) Laboratorio de Genética Humana, Facultad de Medicina/CRIB, Albacete Introducción: La glucoquinasa tiene un papel fundamental en la fisiología de la célula b pancreática, regulando la secreción de insulina en función de los niveles de glucemia. Las mutaciones inactivadoras en hetercocigosis del gen de la glucoquinasa dan lugar a la diabetes MODY 2 (maturity-onset diabetes of the young), de las que se han descrito un número creciente en los últimos años. Caso clínico: Presentamos el caso de un paciente varón de 4 años que presenta glucemias capilares en ayunas >126 mg/dl y postpandriales >140 mg/dl e inferiores a 200 mg/dl. No refiere síntomas cardinales de diabetes mellitus. Antecedentes personales sin interés. Entre los antecedentes familiares destacan padre y tía paterna en estudio por hiperglucemia, y abuelo paterno diagnosticado de diabetes mellitus desde los 70 años en tratamiento actualmente con antidiabéticos orales. No antecedentes de sobrepeso u obesidad en ningún caso. En la exploración física el paciente presenta peso dentro de la normalidad sin evidenciarse otras alteraciones patológicas. En las pruebas comlementarias se objetiva hemoglobina glicosilada de 5,7%, con anticuerpos anti-islotes y anti-GAD (descarboxilasa del ácido glutámico) negativos y péptido C basal de 0,28 ng/ml. Ante la sospecha de diabetes monogénica, se solicita estudio genético familiar, donde se encuentra la mutación, no descrita previamente, V305E en heterocigosis en el exón 8 del gen GCK (figura 1). La importancia del diagnóstico precoz radica en evitar tratamientos crónicos e innecesarios, clasificar correctamente a ambos progenitores y poder llevar a cabo un seguimiento clínico y analítico de estos pacientes y sus descendientes. La historia natural o evolución va a depender del gen o genes afectados y, aunque inicialmente se ha tratado de asociar un tipo de diabetes a una clínica y evolución, lo más importante es el seguimiento clínico y analítico personalizado del paciente.
P1d2d3-062 UNA DELECCIÓN EN EL GEN QUE CODIFICA LA GLUCOKINASA (GLK), CON UNA EXPRESIÓN CLÍNICA MÁS AGRESIVA. L. Regueras Santos (1), A. Diaz Moro (1), E.Hierro Delgado (1), P. Lobo Martinez (1), J.Martinez Saenz de Jubera(1), C. Cizera (2) (1) Servicio de Pediatría del Complejo Asistencial Universitario de León, (2) Instituto de Medicina Molecular, departamento de medicina de la Universidad de Salamanca INTRODUCCIÓN: CASO CLÍNICO: Niña de 7 años remitida a la consulta de endocrinología infantil por presentar, en una analítica en ayunas, una glucemia de 130 mg/dl, con clínica en los últimos dos meses de polidipsia y poliuria sin perdida de peso. En la exploración física presenta un IMC en P75-85 con un estadio de Tanner de 1. Antecedentes familiares: Padre hiperglucemias no estudiadas, abuelo y bisabuelo paterno DM tipo 2, Tío paterno diagnosticado de DM tipo 1 con retinopatía y primo paterno DM tipo 1 que ha precisado Insulina sc Antecedentes personales: Embarazo controlado parto eutócico a término con PRN y LRN adecuados para la edad gestacional. Exploraciones complementarias: Perfil glucémico: glucemias en ayunas entre 80-145 mg/dl y dos horas tras la ingesta entre 120-300 mg/dl, HbA1c 6%. Curva de sobrecarga oral de glucemia: glucosa basal 99 mg/dl y a las 2 horas 333 mg/dl e Insulina basal 3,46 ng/ml. Indice HOMA 0,84; Acs anti IA2 0,1 U/ml, Acs anti-insulina <0,1 U/ml, Acs antiGAD 1,7 U/ml. Función tiroidea T4 libre 1,26 ng/100, TSH 9,65 mcU/ml con anticuerpos antitiroideos negativos. Serología celiaca negativa. Microalbuminuria negativa. Estudio genético paciente: Mutación en el exon 7 del gen GCK en heterocigosis, delección de 32 pares de bases con inserción de 16 pares de bases: N158-159 del ins CCCCTGCTCCTCCTAC. Gen HNF1 sin mutaciones. Estudio genético primo paterno misma mutación. Pendiente estudio genético del padre Evolución: Se recomienda inicialmente sólo dieta, sin observarse cambios en el perfil glucémico ni en la HbA1c, por lo que se inicia tratamiento con Metformina con una respuesta favorable, manteniendo glucemias dentro del rango normal. COMENTARIOS: Esta delección e inserción en el gen de la GCK, no descrita previamente, tiene en nuestro paciente una expresión clínica marcada, presentando datos analíticos de diabetes franca y precisando tratamiento con antidiabéticos orales
P1d2d3-063 DIABETES NEONATAL PERMANENTE POR MUTACIÓN EN GEN DE LA INSULINA. P. Belda Benesiu, P. Díaz Fernández, G. Lou Francés, M. Ferrer Lozano, M. Rodríguez Rigual Hospital Infantil Miguel Servet, Zaragoza La diabetes neonatal es un tipo de diabetes monogénica con dos subgrupos clínicos: transitoria y permanente. La forma permanente requiere tratamiento insulínico contínuo desde el diagnóstico, siendo la causa más frecuente las mutaciones en el gen KCNJ11 que codifica la subunidad Kir6.2 del canal k-ATP de la célula beta pancreática. Caso clínico: niña de 2 meses de edad consulta por fiebre (38,8ºC rectal) de 24 horas de evolución, irritabilidad y rechazo de tomas. No refieren otra sintomatología. Estado general afectado y deshidratación estimada de un 5%. Respiración tipo Kussmaul. A su llegada: glucosa 1010 mg/dL; urea 49 mg/dL; Na/Cl: 128/96 mEq/L; K. 5,3 mEq/L; Osmolaridad: 312 mOsm/L; pH 7,20; pCO2: 13; EB: -22. Glucosuria: 56,46g/L. Además: HbA1c 12,1%; Péptido C: 0,1 ng/mL. Ingresa en UCI inicialmente y una vez superada la cetoacidosis se implanta ISCI. Posteriormente se reciben: amilasa, lipasa y elastasa fecal sin alteraciones; Triglicéridos: 158 mg/dL; LDL/HDL: 117/50 mg/dL. Anticuerpos AntiGAD e IA2 negativos. Control del Péptido C: 0,34ng/mL. Antecedentes personales y familiares: embarazo controlado normoevolutivo, parto eutócico a las 40 semanas. Peso al nacer: 3130g. Screening neonatal negativo. Lactancia materna, buena ganancia ponderal. Padre: hipertrigliceridemia familiar. Madre: tiroiditis autoinmune y bocio multinodular. Diabetes gestacional en el primer embarazo hace 10 años. No consanguinidad. Hermanos sanos. Abuela paterna DM2, fallecida por mieloma. El estudio genético de la niña mediante secuenciación del gen de la insulina (INS) mostró una mutación en el exón 3 en heterozigosis consistente en sustitución de Arginina en posición 89 por Cisteína (c.265 C>T), ya descrita previamente como asociada a diabetes neonatal permanente. Este mismo estudio no encontró anomalías en los padres. Con 7 meses de edad ingresó por descompensación metabólica en contexto de bronquiolitis VRS positivo con buena evolución. Actualmente toma alimentación complementaria, y sigue controles periódicos para ajustar la infusión de insulina en la ISCI según necesidades (en los últimos 3 meses HbA1c: 7.6%, niveles bajos de Péptido C). Discusión. El caso que presentamos reúne las características comunes descritas en las mutaciones del gen INS en heterozigosis: diagnóstico antes de los 6 meses de edad, déficit severo de la función de la célula beta al debut, anticuerpos AntiGad/ICA no detectables, Péptido C bajo o indetectable (basal o postestímulo) y ausencia de clínica extra-pancreática.
P1d2d3-064 HIPOGLUCEMIAS COMO MANIFESTACIÓN DE PREDIABETES TIPO 1 E. Martín Campagne (1), C. Roa Llamazares (2), Mª J. Ballester Herrera, (1), P. Atance, Enrique (1), Sánchez Fernández, Mª José (1) (1) Endocrinología Pediátrica. Hospital General Ciudad Real. (2) Endocrinología y Nutrición. Hospital Santa Bárbara. Puertollano. Ciudad Real Introducción: La hipoglucemia puede ser un síntoma precoz en la diabetes mellitus tipo 2. Se postula que la hiperglucemia puede provocar un hiperinsulinismo compensador postpandrial en las primeras fases de la enfermedad. En la diabetes mellitus tipo 1 se ha descrito una causa de hipoglucemia poco frecuente, la mediada por anticuerpos contra insulina. Se presenta el caso de un varón de 13 años remitido a consulta para estudio de hipoglucemias de repetición. Caso clínico: Varón de 13 años de edad que desde hace un año refiere episodios de clínica adrenérgica (temblor, sudoración y palpitaciones) y neuroglucopénica (mareo y visión borrosa) que ceden con la ingesta de hidratos de carbono. En ocasiones se han objetivado valores de glucemia capilar entre 40-50 mg/dl. Los episodios son de predominio postpandrial. No consumo de fármacos ni tóxicos potencialmente hipoglucemiantes. No cuadro constitucional. El paciente no presenta antecedentes personales de interés. Su hermano mayor padece diabetes mellitus tipo 1 desde los diez años. Exploración física anodina. Pruebas complementarias: analítica: hemograma normal; bioquímica: glucosa 94 mg/dl; creatinina 1,0 mg/dl; colesterol 185 mg/dl; GOT 29 IU/l; GPT 48 IU/l; GGT 51 IU/l; TSH 2,8 mU/ml; HbA1c 4,3%; anticuerpos IA2 positivo 20,2 U/ml; GAD < 0,5. El paciente no acude a posteriores revisiones. Tres años después, es diagnosticado de diabetes mellitus tipo 1, tras semanas de clínica cardinal. Conclusiones: Se presenta el caso de un sujeto con hipoglucemias reactivas en el contexto de prediabetes. Durante el estudio se descubrió la presencia de autoinmunidad contra célula beta pancreática. En la diabetes tipo 1, la aparición de hipoglucemia en estadios iniciales es rara y su etiología es poco clara. Podría estar causada por disfunción de la célula beta o por interacción de anticuerpos anti-insulina. En la actualidad, la determinación de autoinmunidad pancreática sólo tiene valor predictivo, ya que no existe ningún tratamiento preventivo eficaz en la diabetes tipo 1, por lo que no estaría indicada su realización de forma sistemática. En este caso la positividad de los autoanticuerpos permitió orientar la etiología de las hipoglucemias y evitar otras exploraciones innecesarias.
P1d2d3-065 HLA EN POBLACIÓN PEDIÁTRICA CON DIABETES Y. Nóvoa Medina (1), A. Wagner Sahlin (2), O. Montes Ares (3), A. De La Cuesta (1), A. Domínguez (1), S. Quinteiro (1) Complejo Hospitalario Materno Insular de Canarias: (1) Servicio de Endocrinología Pediátrica; (2) Servicio de Endocrinología, (3) Servicio de Inmunología. El Sistema Mayor de Histocompatibilidad (HLA) representa el 40% de la base genética que influye en el desarrollo de la DM1. A continuación presentamos los primeros datos de caracterización del HLA II en una población pediátrica con DM1. Material y Métodos Se realizó tipaje HLA de 62 pacientes diagnosticados de DM1 (según criterios de la ADA), mediante extracción de ADN genomico ( QIAamp, Qiagen) y tipaje HLA-DRB1 y DQB1 mediante PCR-SSP (INNOTRAIN, DiagnostikGmbH). Edad media al debut: 5,6 años (1,1 - 11,5). Distribución por sexos: 50% varones y 50% mujeres Objetivos Realizar caracterización HLA DRB1 y DQB1 de pacientes pediátricos con DM1en Gran Canaria, así como valorar su relación con el sexo y edad de debut. Resultados
Conclusiones * Todos los pacientes presentan DQB1*02 ó DQB1*03. * 89,7% de los pacientes presentan DRB1*03 ó DRB1*04 * Los haplotipos más frecuentemente descritos (DRB1*03-DQB1*02 y DRB1*04-DQB1*03) aparecen en el 85,5% de los pacientes. * El haplotipo de mayor riesgo para la aparición de DM1 (DRB1*03/04 – DQB1*02/03 en heterocigosis) aparece en un 29% de los pacientes, y se asocia a un debut temprano (77% de ellos debutan con < 6 años). * Mayor presencia de DRB1*03 que en población con española con DM 1 con debut a edades más avanzadas (Type I Diabetes Consortium).
P1d2d3-066 CASO CLÍNICO: DEBÚT DIABÉTICO EN PACIENTE CON GRANULOMA ANULAR DISEMINADO. P. Capel Hernández, R. Bermejo Arrieta, M.R. Montero Alonso, A.T. Vila Mas, C. Vidal Palacios Hospital Son Llatzer, Mallorca Introducción: Las afecciones dermatológicas se presentan en el 30-70 % de los diabéticos en algún momento de la evolución de la enfermedad. Generalmente aparecen al inicio de la enfermedad pudiendo ser la primera manifestación sistémica. Otras veces pueden preceder por muchos años al diagnóstico de diabetes melitus (DM). El Granuloma Anular (GA) es descrito como una de ellas, especialmente en su forma múltiple. Caso clínico: Niña de 11 años y medio. Ingresa con el diagnóstico de cetoacidosis diabética grave en contexto de debut diabético. Antecedentes familiares: Historia familiar de DM tipo 2 y DM tipo 1 autoinmune en tío materno, sin otros antecedentes de patología autoinmune de interés. Antecedentes personales: Gestación, parto y periodo neonatal normales. RNT+PAEG. Lactancia materna hasta los 4 m. Seguimiento en CCEE Dermatología desde los 2 años de edad por presentar lesiones cutáneas en cara anterior de miembros inferiores. Se diagnostica de Granuloma Anular Diseminado y se realiza seguimiento. Durante el mismo se realiza, a los 5 años de vida, estudio de TSH y curva de glucemia con resultado normal. Desaparición de las lesiones 7 años después de la primera visita. Historia actual: Se ingresa desde el Servicio de Urgencias Pediátricas con una Glucemia capilar de 542 mg/dl, acidosis metabólica (pH 7.05; pCO2 22.8; HCO3 8.2 y EB -22.3 ) y Cetonemia máxima de 6.1mmol/L. Comentario: La lesión de GA es rara en la infancia, siendo sus localizaciones más frecuentes dorso de pies y manos, muñecas y tobillos. Su causa es desconocida y su relación con enfermedades sistémicas permanece poco clara. No obstante dado que en algunos estudios se ha encontrado asociación entre GA y DM, es importante su conocimiento por los pediatras para un diagnostico y tratamiento precoz de posibles comorbilidades asociadas, en especial la DM.
P1d2d3-067 CARACTERÍSTICAS DE LOS DEBUTS DIABÉTICOS TIPO 1 EN UNA NUEVA UNIDAD DE ENDOCRINOLOGÍA INFANTIL (2005-2011) I. Mulero Collantes, R. Izquierdo Caballero, J.C. Hernando Mayor Hospital Universitario Río Hortega, Valladolid Introducción: La incidencia de diabetes tipo 1(DM1) presenta gran heterogeneidad y variabilidad geográfica. Objetivos: Conocer las características epidemiológicas y clínico-analíticas de los debuts DM1 de 0 a 14 años del período 2005-2011 en nuestra área de salud comparando los resultados con datos nacionales e internacionales. Materiales y métodos: Estudio retrospectivo de los debuts DM1 de 0 a 14 años que ingresaron en nuestra unidad de reciente creación (2005-2011). Datos recogidos durante el ingreso, y cifra de HbA1c a los 6 y 12 meses. Análisis: SPSS 15.0. Resultados; 42 casos (17 niños/25 niñas), lo que supone una incidencia aproximada de 18 casos/100.000 habitantes/año. Mayor incidencia en los meses cálidos, siendo mayo el mes con más casos (8). Edad media: 8 años 2 meses. Grupo etario mayoritario: 5 a 9 años (45,2%), sin diferencias por sexos. Edad mínima al diagnóstico: 17 meses. 26,2% tenían antecedentes familiares de DM1 y 42,9% de DM2. Síntomas más frecuentes: poliuria y polidipsia (90,2%), seguidas del adelgazamiento (53,7%), astenia (29,3%), nicturia (17,1%), polifagia y abdominalgia (14,6%). Un 68,8% tenía un IMC≤p50 (25% un IMC≤P25). Dos casos asociaban una T.Hashimoto y otros dos celiaca. El 53,8% presentaba título de Ac anti-GAD positivo y el 22,5% de Ac antipáncreas. Un 47.3% refería síntomatología 15 días antes del diagnóstico e incluso un 40% más de un mes; presentando cetoacidosis diabética (CAD) el 23,8% (sin cambios estadísticamente significativos en la proporción de debuts con/sin CAD a lo largo del periodo estudiado, ni diferencias por edad o sexo). Media de 9,1 días de ingreso. La glucemia capilar media: 479,9±166,7 mg/dl, (rango de 181-967 mg/dl). HbA1c media inicial: 11,61±1,98%. El 16,7% tuvo algún episodio de descompensación desde el debut hasta su última evaluación. HbA1c media a los 6 meses del debut 7,1±1,4% y a los 12 meses 7,4±0,9%. Conclusiones: Alta tasa de DM1, existiendo diferencias con respecto a otras series que describen una mayor incidencia en los meses fríos y discreto predominio masculino. Muchos pacientes presentaban largo período de síntomas previo al diagnóstico y un 23,8% fueron CAD, lo que nos hace plantearnos la necesidad de una mayor divulgación de programas de sensibilización y reconocimiento de esta enfermedad entre la población.
P1d2d3-068 CARÁCTERÍSTICAS CLÍNICO-EPIDEMIOLÓGICAS DE LAS CETOACIDOSIS DIABÉTICAS AL DEBUT DE ENFERMEDAD DURANTE LOS AÑOS 2007-2011 G. Martínez Moya, M. De Toro Codes, C. Santiago Gutiérrez, C. Arenas Bermúdez, J. De la Cruz Moreno Complejo Hospitalario de Jaén INTRODUCCIÓN: La cetoacidosis diabética (CAD) representa la emergencia metabólica más frecuente en pediatría y la causa principal de morbimortalidad en niños con Diabetes Mellitus tipo I. Constituye la manifestación extrema del déficit de insulina y, en ocasiones, la forma de comienzo de la enfermedad. En nuestro país la frecuencia al diagnóstico de enfermedad está entre el 25-40%, siendo más elevada en los niños menores de 5 años. La mortalidad predominantemente se asocia al edema cerebral y sólo una minoría de las muertes se atribuyen a otras causas. OBJETIVO: Conocer el perfil epidemiológico y clínico de los pacientes atendidos en nuestro hospital con CAD al comienzo de la enfermedad. MATERIAL Y MÉTODOS: Se realiza una revisión retrospectiva de los casos de CAD al debut en los últimos 5 años, de Enero de 2007 a Diciembre de 2011. RESULTADOS: Durante este periodo fueron diagnósticados de Diabetes Mellitus tipo I 58 pacientes, de ellos un 43% fueron como CAD. La mayoría de los pacientes tenían una edad comprendida entre los 5 y los 10 años (17 pacientes) y no hubo diferencias de afectación por género. Los principales motivos de consulta fueron: dolor abdominal y/o vómitos (40%), síntomas cardinales de diabetes (poliuria, polidipsia y pérdida de peso) (32%), decaimiento (16%) y fiebre (12%). El diagnóstico de CAD severa fue de 36% según niveles de pH (pH < 10) y 28% según niveles de bicarbonato (Bicarbonato < 5). Un 24% de nuestros pacientes presentaban deshidratación moderada-severa al ingreso, 16% obnubilación y 8% respiración acidótica. Ninguno de nuestros pacientes presentó complicaciones graves. CONCLUSIONES: - En nuestra casuística la frecuencia de CAD como forma de comienzo de enfermedad es ligeramente superior al descrito, con un predominio de edad comprendido entre los 5 y los 10 años. - Debemos aumentar la formación sobre la enfermedad para intentar disminuir la incidencia de cetoacidosis al debut.
P1d2d3-069 INCIDENCIA DE DIABETES MELLITUS 1 EN UN ÁREA SUBURBANA DE MADRID M.J. Rivero Martín, MªJ. Alcázar Villar, M. Sanz Fernández, D. Montes Bentura, C Navarro Moreno Hospital Universitario de Fuenlabrada, Madrid Introducción: En los últimos años parece haber existido un aumento en la incidencia de diabetes mellitus tipo 1 (DM1), habiendo diferencias entre países, y entre regiones del mismo país. Objetivos: Determinar la incidencia de DM1 en menores de 15 años en nuestro medio. Definir las características epidemiológicas de los pacientes menores de 15 años con debut de DM1. Material y métodos: Estudio prospectivo descriptivo de los pacientes de 0 a 15 años con debut de DM1 desde el 1 de enero de 2008 al 31 de diciembre de 2011 y con residencia habitual en el área de influencia del Hospital Universitario de Fuenlabrada (municipios de Fuenlabrada, Humanes y Moraleja de Enmedio). Los datos de población se recogen de los datos del censo del INE (Instituto Nacional de Estadística). La población de los 3 municipios de influencia a fecha 1 de enero de 2010 era de 38.758 niños de 0 a 15 años. Resultados: La incidencia acumulada para los 4 años de estudio fue de 23,9 casos nuevos/100.000 menores de 15 años. En la tabla 1 se expresa la incidencia por edades. La evolución por años fue 25,8 en 2011, 20,6 en 2010, 25,8 en 2009, y 20,6 en 2008 (sin diferencias significativas). Edad media al diagnóstico: 7,8 años (DS: 3,7). Hemoglobina glicada media al diagnóstico: 13,4 (DS: 2,7). La distribución del debut según el trimestre del año fue: 35% 1º; 11% en 2º; 32,5% en 3º y 21,5% en 4º. 29 (78,4%) eran hijos de familias originarias de España y el 21,6% de familias inmigrantes Discusión: La incidencia en nuestra área de influencia es mayor que la referida para el conjunto de Madrid entre 1997 y 2005, y de las más altas de España. La estacionalidad se mantiene sin cambios respecto a estudios previos. Puede haber pacientes que hayan sido diagnosticados y tratados en otros centros, lo que aumentaría la incidencia.
P1d2d3-070 RESERVA PANCREÁTICA Y PERIODO DE REMISIÓN EN NIÑOS Y ADOLESCENTES CON DIABETES MELLITUS TIPO 1 M. Martín Frías, B. Cano Gutiérrez, P. Enes Romero, R. Yelmo Valverde, M. Alonso Blanco, R. Barrio Castellanos Unidad Diabetes Pediátrica. Servicio de Pediatría. Hospital Universitario Ramón y Cajal. Universidad de Alcalá. Madrid Introducción. La presencia de reserva pancreática tras el diagnóstico de DM1, exponente del grado de destrucción de la célula-b, se relaciona con mejor control metabólico y protección frente a complicaciones agudas y crónicas. Objetivos. Evaluar la función residual de la célula-ß y relacionarla con las características clínico-analíticas iniciales de la DM1. Pacientes y métodos. Estudio prospectivo en 56 pacientes pediátricos con DM1. Analizamos al diagnóstico: edad, pérdida peso (Kg), síntomas cardinales diabetes (meses), alteración metabólica (pH, hiperglucemia/cetosis/acidosis) y HbA1c (%, HPLC-Menarini). Al 1-2 meses evaluamos la reserva pancreática mediante test de glucagón (15µg/kg, máximo 1mg) determinando péptido-C basal y 6 minutos (reserva >0,9ng/ml). Analizamos necesidades insulina (U/Kg/día, remisión <0,5U/kg/día) y HbA1c al mes y/o 6 meses. Estudio estadístico con programa SPSS, versión 17.0; datos expresados en porcentajes, mediana y rango intercuartílico (percentiles 25-75). Resultados. Edad media al diagnóstico 9,5 años (4,7-12,7), 56% impúberes y 52% varones. El 46% mantenían reserva pancreática, sin diferencias entre sexos. Los pacientes sin reserva fueron más jóvenes y tuvieron mayor alteración metabólica al debut (p<0,05). Los pacientes con mayor respuesta de péptido-C mantuvieron menores necesidades de insulina y mejor control metabólico (p<0,05). Conclusiones. La menor edad y la mayor alteración metabólica al debut se asocian con menor reserva pancreática. La persistencia de reserva pancreática facilita el control de la diabetes.
P1d2d3-071 SEGUIMIENTO EVOLUTIVO DE NIÑOS Y ADOLESCENTES CON DIABETES TIPO 1 EN UN HOSPITAL TERCIARIO DE REFERENCIA PARA LA COMUNIDAD N. Lecumberri García, M. Chueca Guindulain, S. Berrade Zubiri, A. Sola Mateos, M.J. Goñi Iriarte, M. Oyarzabal Irigoyen Complejo Hospitalario de Navarra La atención al niño con DM-1 implica prevenir y evitar la aparición de complicaciones agudas y crónicas y detectar asociada (sobre todo autoinmune). La terapia optimizada (MDI, ICSI) es el tratamiento de elección. OBJETIVOS
MATERIAL Y MÉTODOS
RESULTADOS Entre 1990-2011 debutaron 311 niños y adolescentes en Navarra. De ellos, 172 ³17 años (98V, 74M). Edad 23,2 años (rango: 17 - 36). Tº evolución 13,07 años±5DE. HbA1c de toda la vida 8,04% (rango 5,9-12,5). El 94,2% de los pacientes realizan terapia optimizada (MDI y/o ICSI). 71 casos presentaron al menos una complicación aguda (59% HG severa y 41% CAD). Hay diferencias significativas para la HbA1c y el tiempo de evolución (p<0,001) en el grupo con complicaciones. Complicaciones crónicas:25 (14,6%) presentan Retinopatía (simple98.8% y proliferativa 1,2%), 22 (12,8%) Nefropatía (microalbuminuria persistente 3,5% y proteinuria 1,7%) y Neuropatía periférica 6,4%. Hay diferencias significativas con el peor control metabólico y más años en la retinopatía (p<0,001) y nefropatía (p=0,0039), no habiéndolas para la neuropatía. 17% del total asocian al menos otra enfermedad autoinmune (12 celiaca y 43 tiroidea). CONCLUSIONES
P1d2d3-072 DIABETES MELLITUS TIPO 1 CON HEPATITIS AUTOINMUNE E HIPERTIROIDISMO. J.A. Nieto Cuartero, A. Rodríguez Minguez, T. Cabrera, S. Vila Dupla, A. Sillero Sánchez, J. Otero de Becerra Endocrinología, Hospital Infantil Universitario Niño Jesús, Madrid Introducción: La incidencia del hipertiroidismo es del 1% de los niños con diabetes mellitus y de la HAI es del 0,5%. En nuestra experiencia de 300 casos de diabetes mellitus tipo 1 revisados durante 20 años, sólo hemos tenido un caso de diabetes asociado a hipertiroidismo. Lo habitual es que la hepatitis se de en primer lugar. Nuestro caso es excepcional ya que primero fue la diabetes y luego la hepatitis. Material y Métodos: Mujer que desde los 10 años y 3 meses de vida presenta diabetes mellitus tipo 1, bien controlada hasta el momento actual, con hemoglobina glicosilada A1C del 7,1%, sin microalbuminuria y con perfil lipídico normal. HLA DR3-DR4. Creatinina 0,93 mg/dL. Anticuerpos antitiroideos negativos. Desde el 11 de febrero del 2008, se detecta hepatitis autoinmune con marcada elevación de GOT, GPT, yGGT, IgC, gammaglobulinas y + para ANA, AML y AntiLKM, F-actina, antiLC-1 y anticélulas parietales.BH: Hepatitis crónica activa. Se inicia tratamiento con esteroides a dosis de 30 mgrs/dia e azatrioprina a dosis de 100 mgrs. En febrero del 2012 la GOT la GPT y la GGT son normales. La T4 libre actual elevada 2,3ng/dl, la TSH baja 0,22 mul/ml. Se instauró tratamiento con antitiroideos. A reseñar la aparición de edemas en miembros inferiores secundarios a hipoalbuminemia, que cedieron tras la administración de concentrados de proteínas diarias. En la actualidad la talla de de 1,63 m, tensión arterial normal 116/73, 58 Kg, fondo de ojo: normal, rendimiento escolar bueno, se mantiene con insulina diaria de glargina 2 inyecciones diarias más suplementos de insulina regular cuando lo precisa. Los autocontroles de glucemia diarios son muy aceptables Resultados: Reseñamos que la instauración precoz del tratamiento en el caso de nuestra paciente ha normalizado la función hepática beneficiándose del tratamiento combinado de corticoides y azatioprina ajustándose con la dosificación de la TPMT, tras 4 años la evolución es excelente, como lo demuestran los estudios hepáticos e inmunológicos. Conclusiones: 1. Reseñar que la combinación de esteroides y azatioprina, ha sido excelente en cuanto al control de la hepatitis autoinmune. 2. La coexistencia de hipertiroidismo con la diabetes no ha empeorado el control de la diabetes.
P1d2d3-073 DESCRIPCIÓN DE LOS PACIENTES DIABÉTICOS TIPO 1 EN EL HOSPITAL COMARCAL DE REFERENCIA E. Farreny Sastre (1), G. Martí Aromir (1), S. Burgaya Subirana (2), E. Abadal Soler (3), J. Sitjes Costas (2), S. Nevot Falcó (2) (1) Servicio de Endocrinología pediátrica; (2) Servicio de Pediatría; (3) D.U.E. Educadora en diabetes. ALTHAIA. Xarxa Hospitalària de Manresa. Hospital Sant Joan de Déu. Introducción: Las Consultas Externas (CCEE) de Endocrinología Pediátrica de nuestro hospital han ido creciendo en los últimos años con el control y seguimiento de los pacientes diabéticos tipo 1 (DM1). Objetivos: Determinar las características del debut y seguimiento de los DM1 controlados en CCEE de Endocrinología Pediátrica de un hospital comarcal hasta el año 2011. Material/métodos: Estudio descriptivo retrospectivo de 32 pacientes DM1 con edades entre 0 y 15 años. Resultados: Se analizaron 32 pacientes (51,3% varones). Debut: El 87,5% debutaron en nuestro hospital. La edad media fue de 7,2 años (rango 1,4-14,7 años). Distribución por edades: 0-5 años: 31,2%, 5-10 años: 40,6%, 10-15 años: 28,1%. El 78,1 % debutaron en estadio prepuberal. 84,4% raza caucásica y 15,6% magrebí. El 56,7% presentaron cetoacidosis (CAD). Tiempo medio de evolución de los síntomas: 23,6 días. HbA1c media de 12,2%. Dosis media de insulina 0,8 U/kg/día. El 76% tenían al menos un marcador de inmunidad de DM1 positivo (AAI, GAD, ICA o IA2). El 20,8% tenían anticuerpos antitiroideos positivos y el 17,2% anticuerpos antitransglutaminasa positivos. 6,2% presentaban vitíligo. Antecedentes familiares: 34,4% DM2, 28,1% DM1, 6,2% diabetes gestacional, 6,2% patología tiroidea, 3,1% celiaquía, 3,1% vitíligo, 3,1% enfermedad de Crohn. Seguimiento: Edad media actual 10,8 años. Tiempo medio de evolución 3,6 años. HbA1c media post-debut 7,9% y en el último año 7,8%. Dosis media de insulina 0,7 U/kg/día. Tipo de insulina basal: 84,4% glargina y 15,6% detemir. 34,4% asocian insulina regular y 65,5% análogos de acción rápida. 40,6% utiliza mezclas. 12,5 % tiroiditis autoinmune y 6,2% vitíligo. No diagnósticos de celiaquía. El 85,7% acepta su enfermedad, 88,9% realiza él los autocontroles y 66,7% se autoadministra la insulina. 56,2% realiza deporte. Conclusiones: Mayor frecuencia del debut entre los 5 y 10 años. Más de la mitad de los pacientes debutaron en forma de CAD y el tiempo de evolución de los síntomas es elevado. Se debe insistir en las campañas de detección precoz de la diabetes. Con las pautas actuales de insulinoterapia conseguimos un control metabólico aceptable. Consideramos positiva la aceptación de la enfermedad así como el grado de autonomía del paciente dependiendo de la edad.
P1d2d3-074 UTILIDAD DEL COCIENTE TRIGLICERIDOS-HDLCOLESTEROL PARA IDENTIFICAR FACTORES DE RIESGO CARDIOVASCULAR EN ADOLESCENTES CON DIABETES TIPO 1 DE LARGA EVOLUCIÓN L. Golmayo Gaztelu, P. Ros Pérez, M.Martín Frías, M.Alonso Blanco , E. Colino Alcol, R. Barrio Castellanos Unidad de Diabetes Pediátrica. Hospital Universitario Ramón y Cajal. Universidad de Alcalá. Madrid
Introducción: La diabetes tipo 1 (DM1) constituye un factor de riesgo independiente para la enfermedad cardiovascular, asociando una elevada morbi-mortalidad. El cociente Triglicéridos/HDL-colesterol (TG/HDLc) está estrechamente relacionado con el riesgo cardiovascular y metabólico en adultos, independientemente de la obesidad. Estudios recientes en la edad pediátrica han demostrado que un TG/HDLc elevado (≥ 2) se asocia a un perfil cardiovascular desfavorable, pero no hay estudios al respecto en población pediátrica con DM1. Objetivo: Estudiar la relación entre el cociente TG/HDLc y varios factores de riesgo cardiovascular (FRCV) en adolescentes con DM1 de larga evolución. Pacientes y métodos: Analizamos la relación entre el cociente TG/HDLc y determinados FRCV en 99 pacientes (55% varones), a los nueve años del diagnóstico de DM1 (tiempo medio de evolución 9,2±1,3 años). Consideramos los siguientes FRCV: grado de control metabólico (media de HbA1c del año previo (Menarini; HPLC;VN 5,31%±0,21), IMC ( Hernández 1988),dosis de insulina (u/Kg/día), colesterol total (CT), triglicéridos (TG), LDLc, HDLc, excreción urinaria de albúmina (EUA) y tensión arterial (TA). Se definió HTA como TA>p90 (Task Force 2004). Los pacientes fueron clasificados en 3 grupos según el cociente TG/HDLc bajo (<1,2); intermedio (1,2 a 2) y alto (≥2). El análisis estadístico se realizó mediante SPSS. Los datos aparecen expresados en medias ± DE y medianas con rangos. Las medias y medianas fueron comparadas mediante ANOVA y K-Wallis, respectivamente. Resultados: Ver tabla. No se observaron diferencias significativas en el IMC, HTA ni EUA entre los grupos. Conclusiones: Un TG/HDL-colesterol elevado (≥2) en pacientes adolescentes con DM1 de larga evolución podría ser un marcador útil para la identificación de pacientes con peor perfil cardio-metabólico.
P1d2d3-075 VITAMINA D Y METABOLISMO HIDROCARBONADO EN PACIENTES CON FIBROSIS QUÍSTICA EN EDAD PEDIATRICA M. Martín Frías (1), A. Lamas Ferreiro (2), B. Cano Gutiérrez (1), P. Enes Romero (1), R. Yelmo Valverde (1), R. Barrio Castellanos (1) (1) Unidad de Diabetes Pediátrica, Servicio de Pediatría Hospital Universitario Ramón y Cajal, Universidad de Alcalá, Madrid (2) Unidad de Fibrosis Quística. Servicio de Pediatría, Hospital Universitario Ramón y Cajal, Universidad de Alcalá, Madrid Introducción. El déficit de vitamina D se ha relacionado con el desarrollo de diabetes en la población general. En los pacientes con fibrosis quística (FQ) es frecuente tanto la presencia de niveles subóptimos de vitamina D como de alteraciones hidrocarbonadas (AH). Objetivos. Analizar los niveles de vitamina D en pacientes con FQ en edad pediátrica y su posible relación con la presencia de AH. Pacientes y métodos. Estudio prospectivo en 38 pacientes pediátricos con FQ. Se determinaron anualmente los niveles de vitamina D (VN >20ng/ml) y la respuesta glucémica a sobrecarga oral de glucosa (SOG) realizada en fase estable de enfermedad, hasta objetivar presencia de AH; analizamos un total de 65 SOG. Todos los pacientes recibían suplementos con dosis estándar de vitamina D según niveles séricos. Analizamos: edad, sexo, presencia y tipo de AH y HbA1c (HPLC-Menarini). Estudio estadístico realizado con programa SPSS-versión-17.0, pruebas no paramétricas nivel significación p<0,05; datos expresados en porcentaje, mediana y rango intercuartílico. Resultados. Edad media 11,2 años (8,8-14,7), 70% varones, 54% púberes. Niveles medios de vitamina D 21,3ng/ml (17,8-27,3), teniendo el 38% de los pacientes niveles deficitarios, independientemente de la edad y del sexo. En el 80% de los pacientes se detectó AH, diabetes en el 18%. De los 8 pacientes que desarrollaron AH en el seguimiento, 4 empeoraron niveles de vitamina D respecto al año previo (media de -13,4ng/ml); en el resto mejoría media de +3ng/ml. No encontramos diferencias significativas entre las variables analizadas en función de la existencia o no de déficit de vitamina D. Tampoco encontramos niveles inferiores de vitamina D en los pacientes con diabetes. No detectamos correlación significativa de vitamina D con HbA1c ni con glucemia a los 120 minutos de la SOG. Conclusiones. El déficit de vitamina D y la presencia de AH son frecuentes en los pacientes con FQ. No encontramos relación entre los niveles de vitamina D y las alteraciones del metabolismo hidrocarbonado.
P1d2d3-076 SÍNDROME DE MAURIAC EN PACIENTES AFECTOS DE DIABETES MELLITUS TIPO I DE LARGA EVOLUCIÓN Y MAL CONTROL METABÓLICO; REVISIÓN DE CUATRO CASOS A. Campos Martorell, B. Valle del Barrio. M.A. Albisu Aparicio, M. Gussinyé Canabal, D. Yeste Fernandez, A. Carrascosa Lezcano Hospital Universitari Vall d Hebron, Barcelona FUNDAMENTO Y OBJETIVOS Se ha descrito en pacientes afectos de diabetes mellitus tipo I (DMI) con mal control metabólico, hepatomegalia por depósito de glucógeno intrahepático e hipertransaminemia. Si estos pacientes asocian dislipemia, rasgos cushingoides y retraso del crecimiento o del desarrollo podemos hablar de Síndrome de Mauriac (SM). En las últimas décadas con las pautas intensivas con nuevas y mejores insulinas, este síndrome se ha convertido en una rareza. La importancia de este cuadro radica en que constituye la causa más frecuente de disfunción hepática en niños con DMI y que es reversible mediante la optimización de la insulinoterapia. El objetivo de este trabajo es presentar cuatro pacientes visitados en nuestro hospital en los últimos diez años, con criterios diagnósticos de SM, describir sus características clínicas, metabólicas, analíticas y las complic MATERIAL Y MÉTODOS Presentamos 4 adolescentes (dos mujeres y dos varones) con DMI de larga evolución y mal control metabólico, edades comprendidas entre los 13 y los 14 años, que presentaron hepatomegalia, hipertransaminemia y dislipemia con funcionalismo hepático normal. En uno se diagnosticó pancreatitis leve. Todos ellos presentaron retraso de crecimiento, uno de ellos muy severo y otro hipogonadismo hipogonadotropo. Tres de los cuatro pacientes ingresaron en el servicio de urgencias por descompensación metabólica (dos presentaron cetosis y uno cetoacidosis) siendo el cuarto paciente remitido directamente a la consulta de endocrinología. En todos los casos la hemoglobina glucosilada al ingreso fue superior a 9% y las ecografías hepáticas objetivaron aumento del tamaño hepático con alteración de la ecogenicidad sugestiva de acumulación de glucogéno intrahepático. Los cuatro pacientes presentaron una correcta evolución clínica tras la optimización de la insulinoterapia. CONCLUSIONES:
P1d2d3-077 VARIABILIDAD CLÍNICA EN EL USO CON ISCI DE GLULISINE VS ASPART/HUMALOG EN NIÑOS DM TIPO1. OBSERVACIÓN POR EVENTO FARMACÉUTICO INESPERADO I. Díez López, A. Sarasua Miranda, I. Lorente Blázquez Sección Endocrinología Infantil, Hospital Universitario de Álava. Sede Txagorritxu. Vitoria Introducción: La composición química de los diferentes análogos rápidos, les confiere peculiaridades farmacocinéticas-dinámicas. Destaca la ausencia de zinc de glulisine, otorgándole una menor cristalización, posible ventaja en dispositivos con catéter. No existen estudios previos sobre esta peculiaridad en pacientes pediátricos con ISCI. De Octubre a Diciembre de 2011 existe un evento farmacéutico inesperado interrumpe la distribución de glulisine a nivel mundial. Todos los pacientes usuarios durante este tiempo deben de pasar a otro análogo de insulina rápida. Objetivo: Evaluar el impacto de este evento, y por lo tanto el cambio de insulinas en el control metabólico y calidad de vida de los niños con ISCI Métodos: niños DM tipo 1a, con al menos 36 meses de evolución desde su début y con 12 meses de uso de ISCI con insulina glulisine hasta Oct.2011. Estudio variables HbA1c (DCA), glucemias preprandiales(GPre), postprandiales(Gpos), necesidades insulina(NI), días uso catéter(DC) y encuesta satisfacción global (1-10)(Enc). Cambio obligado a aspart/humalog y revaluación en Enero 2012. Estudio comparativo. SPSS 17.0., muestras pareadas no paramétrico n<30. Resultados: 5 niños (2♂ ) , edad media de 11.5 años [8-13]. Con glulisine y antes del cambio è medias variables HbA1c (DCA): 7.6%[6.8-8.2], Gpre 127 mg/dl[65-185], Gpos 149 mg/dl[98-195], NI: 0.88 UI/kg/día[0.72-1.18], DC:4,3 dias[3.5-5] y Enc 8.5 puntos [7-10]. Tras periodo sin glulisine y paso a aspart/humalog. è medias variables HbA1c (DCA): 8.2%[7.4-9.1] diferencias p:0.02 IC95%[0.4-0.8], Gpre 148 mg/dl[102-190] diferencias p:0.01 IC95%[42-68], Gpos 175 mg/dl[141-245] p:0.04 IC95%[32-76], NI: 1.02 UI/kg/día[0.85-1.31] diferencias p:0.01 IC95%[0.12-0.34], DC:2.6 días[2-3.5] p:0.001 IC95%[1.5-2.5], y Enc 6 puntos [5-7]. Conclusión: A pesar de la potencia del estudio por escasa n los resultados apuntan a las posibles ventajas clínicas y económicas que pueden derivarse del uso de glulisine en este tipo de pacientes. Recomendamos este tipo de insulina en niños DMtipo1 con ISCI.
P1d2d3-078 EFICACIA DEL TRATAMIENTO CON ISCI EN PACIENTES PREPUBERALES Y PUBERALES CON DIABETES MELLITUS TIPO 1 P. Enes Romero, B. Cano Gutiérrez, R. Yelmo Valverde, M. Alonso Blanco, M. Martín-Frías, R. Barrio Castellanos Unidad de Diabetes Pediátrica. Hospital Universitario Ramón y Cajal. Universidad de Alcalá. Madrid Introducción. El tratamiento con ISCI en DM1 parece ser más eficaz en pacientes prepuberales por su mejor adherencia al mismo. Objetivos. Comparar la eficacia del tratamiento con ISCI entre niños prepuberales y puberales con DM1. Pacientes y métodos. Estudio retrospectivo en 59 pacientes con DM1, comparando los 29 prepuberales con 29 puberales de los 64 tratados en nuestra Unidad de Diabetes (64% y 43% varones, respectivamente). Analizamos en el año previo a la ISCI y 4 años posteriores: HbA1c (HPLC-Menarini 5,3±0,41), necesidades insulina (u/kg/día), tramos de basal, glucemias capilares/día, hipoglucemias graves (HG) y cetoaciosis. Estudio estadístico con programa SPSS 20.0, datos expresados mediana y rango intercuartílico. Resultados. El 48 % de los prepuberales y 46% de los puberales redujeron la HbA1c ≥0,2% durante el 1º año de ISCI. La dosis de insulina disminuyó significativamente al inicio en ambos grupos, manteniéndose durante los 4 años. Sólo 2 pacientes (uno en cada grupo) presentaron un episodio de HG con ISCI, ambos con antecedentes de 2 y 3 episodios en al año previo. Un paciente puberal tuvo un episodio de cetoacidosis en relación con consumo de alcohol. Abandonaron la ISCI 4 pacientes puberales y ninguno prepuberal. Durante el primer año, en el grupo puberal existe relación directa entre disminución HbA1c y número glucemias capilares/día (p=0,026). Conclusiones. El tratamiento con ISCI es una alternativa eficaz y segura en niños y adolescentes con DM1. Los beneficios obtenidos en el control metabólico son discretamente mejores en niños prepuberales.
P1d2d3-079 CONTROL METABÓLICO A LARGO PLAZO EN NIÑOS Y ADOLESCENTES CON DIABETES TIPO 1 EN TRATAMIENTO CON INFUSIÓN SUBCUTÁNEA CONTINUA DE INSULINA R. Cardona Hernández (1), L. Suárez Ortega (1), M.Z. Bosch (1), J. Ho (2), C. Mameli (3), A.E. Scaramuzza (3) (1) Hospital Sant Joan de Déu, Sección de Endocrinología, Barcelona; (2) Department of Pediatrics, University of Calgary, (Canada); (3) Luigi Sacco, Department of Pediatrics, University of Milano (Italy) Introducción.- Existen pocos datos acerca del seguimiento a largo plazo del tratamiento con infusión subcutánea continua de insulina (ISCI) en niños y adolescentes con diabetes tipo 1 (DM1). Objetivos: 1) Comparar el control metabólico de niños y adolescentes con diabetes antes del tratamiento con ISCI y durante los 4 primeros años de tratamiento en una Unidad de Diabetes de un hospital terciario. 2) Comparar los resultados obtenidos, en el contexto de un estudio multicéntrico, con los de dos hospitales terciarios de Canadá e Italia. Métodos.- Estudio retrospectivo (revisión de historias clínicas); Criterios de inclusión: edad 4-20 años, DM1 de ≥4 años de evolución, tratamiento con ISCI ≥4 años de duración; Variables: HbA1C, requerimientos insulínicos, IMC, hipoglucemias y episodios de cetoacidosis. Recogida de datos independientemente en 3 países (España, Canadá e Italia). Análisis estadístico: T de Student para datos apareados. Prueba de Kruskall-Wallis para comparativas grupales. Resultados.- 34 sujetos cumplen criterios de inclusión en nuestro centro (22 varones) con edad 4-20 años (14.6±3.8), duración de diabetes 10.6±3.2 años e inicio de la terapia ISCI a los 9.1±4.0 años. Tras el inicio de la terapia ISCI la HbA1C descendió significativamente sólo durante el primer año (a los 6m Δ=-0.72 p=0.021 y a los 12m Δ=-0.72 p=0.037). Posteriormente la HbA1C no mostró diferencias significativas respecto a la inicial. No se detectaron diferencias en los requerimientos de insulina, el IMC, el número de hipoglucemias graves ni en los episodios de cetoacidosis entre el inicio de la terapia y el periodo de seguimiento. Se observaron resultados similares en los centros de Canadá e Italia (n total = 110 sujetos). Conclusiones.- Aunque la terapia ISCI es efectiva a largo plazo el mayor beneficio se obtiene durante el primer año de terapia. Durante el seguimiento a 4 años, los valores de HbA1C tendieron a incrementarse. Es necesario estudiar qué factores condicionan este incremento de HbA1C a partir del primer año.
P1d2d3-080 NECROBIOSIS LIPOÍDICA EN UN PACIENTE PEDIÁTRICO CON DIABETES MELLITUS TIPO 1 C. del Castillo Villaescusa, A. Navarro Ruiz, P. Codoñer Franch Hospital Universitario Dr. Peset. Valencia INTRODUCCIÓN: La necrobiosis lipoídica (NL) es una lesión cutánea producida por degeneración del colágeno caracterizada por placas irregulares con borde eritematoso y centro atrófico amarillo-parduzco, de predominio pretibial. Etiología desconocida y prevalencia variable en niños (0.06-10%), con una fuerte asociación con DM1 (hasta 60% de los pacientes con NL son diabéticos). En ocasiones la lesión precede en años al diagnóstico de diabetes. CASO CLÍNICO: Escolar mujer de 11 años con lesiones cutáneas en cara anterolateral de ambas piernas en región tibial. Antecedentes familiares sin interés. Diabetes mellitus tipo 1 desde los 2 años. Mal control metabólico con HbA1c media en los últimos tres años de 8,3%, con discreta mejoría en el último año (media 7,9%). No complicaciones microvasculares. Epilepsia tratada con valproato hasta los 9 años. Aparición de las lesiones a los 8 años, de curso lento, inicialmente como pequeñas máculas eritematosas que se fueron extendiendo dando bordes irregulares y centro amarillento y atrófico, con telangiectasias en superficie. Biopsia compatible con necrobiosis lipoídica. Tratamiento inicial con pimecrolimus durante un año sin mejoría. Posteriormente crema de rosa de mosqueta y vitamina E asociando a los 6 meses ácido acetilsalicílico oral a bajas dosis. Mejoría de las lesiones existentes con disminución de la consistencia y crecimiento más lento, pero en los últimos meses aparición de nuevas lesiones. CONCLUSIONES: La NL es una patología cutánea asociada a la diabetes, poco frecuente y excepcional en niños. Se han probado múltiples tratamientos en adultos (corticoides tópicos, sistémicos e intralesionales, dipiridamol, ácido acetilsalicílico, nicotinamida, pentoxifilina, tretinoína, ciclosporina o micofenolato, así como láser, oxígeno hiperbárico, factor estimulante de colonias de granulocitos y PUVA. Ninguno ha demostrado ser efectivo en ensayos controlados y la mayoría tienen efectos secundarios significativos. Las lesiones ulceradas dolorosas se han tratado con tacrolimus tópico, cirugía e injertos cutáneos. La aparición y evolución de la NL es independiente de la duración de la diabetes, edad del paciente y control metabólico. En niños puede ser de difícil manejo y se ha referido riesgo a largo plazo de transformación en carcinoma epidermoide.
P1d2d3-081 UTILIDAD DE UNA PAGINA WEB PARA LA EDUCACIÓN SANITARIA DE NIÑOS DIABÉTICOS J.J. Momblan De Cabo, J.L. Gómez Llorente, P. Oliva Pérez, A. Bonillo Perales CHT Torrecárdenas (Almería) Objetivo Plantear la utilidad de las páginas web para la educación sanitaria de niños diabéticos de manera que sean una herramienta útil para obtener y/o refrescar conocimientos sobre su enfermedad Material Se realizó una pagina web, en Mayo 2011, cuya dirección es www.diabetesinfantilcht.com donde se incluían diversos apartados: alimentación, insulinas, ejercicio e insulina, factor de sensibilidad y ratio, noticias y calendarios y cuestionarios. Además de vínculos importantes para diabetes, horario y teléfono de consulta, email de la consulta y cartera de servicio. Para determinar su uso se utilizó tanto un contador de visitas en la primera pagina como los cuestionarios que pondrían ser descargados y contestados por los pacientes y/o familiares y eran recogidos en la consulta. La pagina web en su totalidad se presentaba a los pacientes según demanda a la consulta, que aproximadamente es cada 4 meses, por lo que hasta ahora se ha anunciado a mas del 50% del total de los pacientes, así mismo se presentaba en las sesiones grupales que son mensuales. Los cuestionarios se van actualizando cada 2 meses y consta en de 5 preguntas de respuesta múltiple Resultados El contador de visitas se disparó desde su puesta en marcha hasta mas de 600 y casi todos los pacientes entregaron los cuestionarios en las consultas (cerca del 80%). La satisfacción con dicha pagina se obtuvo por pregunta directa , por respuesta en la última pregunta del cuestionario así como por cuestionario de satisfacción al final de cada educación grupal. Esta satisfacción rondó el 90%. La página ha obtenido dos certificaciones de calidad: web de interés sanitario y web médica acreditada. Conclusiones Creemos que la pagina web de estas características aporta un plus muy importante para el manejo del paciente diabéticos pues puede aclarar muchas dudas o preguntas que le pueden surgir y es un medio de comunicación y adhesión del paciente sobre todo en edad juvenil.
P1d2d3-082 TELEMEDICINA APLICADA A LA DIABETES TIPO 1 EN LA EDAD PEDIÁTRICA: ESTUDIO PILOTO. P. Ros Pérez (1), E. Colino Alcohol (1), A. Ruiz Serrano (1), N. Lacamara Ormaechea (1), M. Martín Frías (2), R. Barrio Castellanos (2). (1) Hospital Universitario Puerta de Hierro-Majadahonda, Unidad de Endocrinología y Diabetes Pediátrica, Madrid; (2) Hospital Universitario Ramón y Cajal. Unidad de Endocrinología y Diabetes Pediátrica, Madrid Introducción. La aplicación de las nuevas tecnologías de la comunicación a la medicina (telemedicina) ha permitido optimizar la atención diabetológica, contribuyendo al mantenimiento del control metabólico. No obstante, la evidencia de su efectividad e impacto en el control metabólico es limitada, sobre todo en la población pediátrica con diabetes tipo 1 (DM1). Objetivo: Evaluar la viabilidad de la monitorización telemática (MT) y su eficacia en el control metabólico de pacientes pediátricos con DM1 en tratamiento intensivo, así como el grado de satisfacción y calidad percibida. Pacientes y métodos: Estudio piloto observacional, descriptivo, sin grupo control. Se incluyeron 32 pacientes: 13 con < 1 año de evolución (grupo 1) y 19 con > 1 año de evolución (grupo 2) (tabla 1). Se encontraban en pubertad (Tanner > III) en 6/13 (grupo 1) y 12/19 (grupo 2). La MT se realizó mediante correo electrónico, fax y teléfono móvil. La frecuencia de contacto mínima fue establecida en el grupo 1 (semanal, quincenal y mensual) y a demanda en el grupo 2, con un mínimo mensual. El grado de satisfacción y calidad percibida se analizó mediante encuesta específica (ESCP). Los valores de HbA1c (Menarini, HPLC; VN:5.3%±0.2), como indicador del control metabólico, se expresan como media ± DE. Resultados. En ambos grupos se objetivó un mantenimiento de los niveles de HbA1c (tabla1). Ninguno de los pacientes tuvo hipoglucemias graves, precisó visitas “extra” ni consultas en urgencias. Se objetivaron descompensaciones (9/32), que se resolvieron con el apoyo MT. Contestaron a la ESCP 31 de los 32 pacientes y todos encontraron la TM útil para mejorar su adherencia al tratamiento y capacidad de autocontrol. Conclusiones 1. La telemedicina constituye una herramienta viable, útil y eficaz para la monitorización y el mantenimiento del buen control metabólico, tanto en el debut como evolutivamente, de los pacientes pediátricos con DM1. 2. Existe gran aceptación de la telemedicina por parte del paciente, favoreciendo su adherencia al tratamiento.
P1d2d3-083 : CALIDAD DE LA DIETA MEDITERRÁNEA Y NIVEL DE ACTIVIDAD FÍSICA EN PACIENTES CON DM1 Y SUS FAMILIAS. L. Sentochordi Montane (1), I. Martínez-Badás (1), V. Sebastián Ibáñez (2), V. M. González González (1), C. Julve Ramos (1), S. López Bravo (1) (1) Servicio Pediatría, H. Universitario Infanta Leonor, Madrid. (2) Unidad de Soporte a la Investigación Clínica, H. Universitario Infanta Leonor. Introducción: La experiencia en el tratamiento de la obesidad infantil nos recuerda la importancia de los hábitos familiares en la consecución de los objetivos del paciente con diabetes tipo 1. La educación diabetológica orienta a las familias hacia un estilo de vida saludable para todos los miembros. Objetivos: 1- Describir en pacientes con diabetes tipo 1 y sus familias la presencia de sobrepeso/obesidad, la calidad de la dieta mediterránea y el grado de actividad física. 2- Compararlo con familias con hijos sin diabetes. 3- Establecer relaciones con los valores de HbA1c de los pacientes con DM1. Material y métodos: Se realizó un trabajo preliminar mediante un estudio transversal a un grupo de pacientes en seguimiento por DM1 en la consulta de endocrinología así como a sus familias. Los controles sanos fueron familias del personal del área de Pediatría. Todos rellenaron un cuestionario con 27 preguntas (4 filiación y antropometría, 16 Indice KidMed, 7 Cuestionario Internacional de Actividad Física-IPAQ). Resultados: Se recogieron datos de 9 familias con un hijo con diabetes tipo 1 y de 11 familias control. Encontramos obesidad en 11% diabéticos, 5,5% padres y 5% madres. Encontramos sobrepeso en 61% padres, 40% madres y 7.6% hermanos. La mediana global del índice Kidmed fue de 8. La mediana en las familias con diabetes fue de 8.5 y en las familias sin diabetes 7.5. La mediana de los pacientes con diabetes fue de 10, siendo la de los hermanos con y sin diabetes de 8. Los pacientes con diabetes obtuvieron mejores resultados en el IPAQ que el grupo de hermanos, padres y madres. No se encontró relación entre la media de HbA1c del año anterior y el índice Kidmed así como tampoco con el IPAQ. Conclusiones: 1. Los pacientes con diabetes tipo 1 y sus familias tienen puntuaciones más altas del índice Kidmed. 2. Los pacientes con diabetes tipo 1 realizan mayor actividad física que sus familias y los controles. 3. No encontramos relación entre índice Kidmed y HbA1c en la muestra de pacientes estudiada.
P1d2d3-084 PERCEPCIÓN DE CALIDAD DE VIDA EN NIÑOS CON DIABETES MELLITUS TIPO 1 S. de la Torre Santos (1), L. Bertholt (2), E. Maldonado Ruiz (1), S. Alberola López (3), I. Pérez García (3), M.C. González Torroglosa (1) (1) Complejo Asistencial de Palencia. Palencia, (2) C.S. Aguilar de Campoo. Palencia, (3) C.S. Jardinillos. Palencia INTRODUCCIÓN: Este trabajo pretende conocer la calidad de vida percibida por los pacientes pediátricos con diabetes tipo 1 y sus padres y compararlos con un grupo de niños sanos. MATERIAL Y MÉTODOS: El estudio incluyó 18 pacientes diabéticos tipo 1 de nuestro área sanitaria, con edades comprendidas entre 4-14 años (mediana 11,5). Se realizó un emparejamiento por edad, sexo y ámbito (rural/urbano) con un grupo control de niños sanos. Se utilizó el cuestionario KINDL-R, que dispone de diferentes versiones para cada grupo de edad: 4-7 años (Kiddy-Kindl), 8-12 años (Kid-Kindl), y 13-16 años (Kiddo-Kindl). Además incluye dos versiones para los padres de 4-7 años y de 8-16 años. Contiene 24 preguntas distribuídas en 6 dimensiones: bienestar físico, bienestar emocional, autoestima, familia, amigos y colegio. Contiene un módulo adicional dirigido a niños con enfermedades crónicas. En el grupo de diabéticos se anotó el tiempo de evolución de la enfermedad y la cifra de hemoglobina glicosilada (HbA1C). RESULTADOS: La distribución por sexos fue de 56% varones y 44% mujeres. El 72% de los niños reside en medio urbano. Al comparar las 6 dimensiones entre el grupo de casos frente a los controles tanto en los niños como en los padres, no se han detectado diferencias estadísticamente significativas. La comparación en la percepción de los padres frente a sus hijos únicamente mostró diferencias estadísticamente significativas (p=0,018) en la dimensión colegio. No se encontraron diferencias en el cuestionario de calidad de vida al comparar entre sexos y tiempo de evolución de la enfermedad. Al realizar las comparaciones en función de la cifra de HbA1C se encontraron diferencias estadísticamente significativas a favor del grupo con mejor control metabólico en las siguientes dimensiones: Niños: calidad de vida total (p=0,02), colegio (p=0,03), enfermedad crónica (p=0,03). Padres: calidad de vida total (p=0,04). CONCLUSIONES:
P1d2d3-085 COMPLICACIONES NEONATALES Y OBSTÉTRICAS EN LOS HIJOS DE MADRE DIABÉTICA. RELACIÓN CON EL TIPO DE DIABETES Y LA HEMOGLOBINA GLICOSILADA B. Bautista, P. González, P. Prieto Matos, C. Lamas, T. Carbajosa, J. Prieto Veiga Hospital Universitario Salamanca INTRODUCCIÓN: La diabetes mellitus (DM) gestacional es la complicación más frecuente de las gestantes. Se define como aquella DM que se identifica por primera vez durante la gestación. Su importancia radica en que está cualquier tipo de DM aumenta el riesgo de complicaciones obstétricas e incluso que la DM aumenta el riesgo de malformaciones. PACIENTES Y MÉTODOS: Se realiza un estudio retrospectivo, observacional eligiendo al azar historias de hijos de madre diabética. Se recogen datos sobre la gestación, la antropometría neonatal, analítica neonatal y materna, el parto, y malformaciones del neonato. Se realiza análisis estadístico con el programa SPSS 17. RESULTADOS: Se recogen 138 pacientes con una edad gestacional de 36,6±2,8 semanas. El diagnóstico materno en el 75% es DM gestacional, el 18,8% DM tipo 1, el 4,3% DM tipo 2 y el 1,4% otro tipo. El 87,5% de las DM gestacionales se trataron con dieta. La Hb1Ac materna es 5,68±1,2%. Se demuestran diferencias estadísticamente significativas entre la Hb1Ac de DM 1 con respecto al resto de diagnósticos (p =0,000), no encontrándose diferencias entre el resto de grupos entre sí. El 57,1% de los partos son cesáreas y el 13% inducidos. El 93,4% de los neonatos ingresaron y la duración del ingreso fue de 10,82 ± 12,4 días. Tras el embarazo la DM desapareció en el 92,3% de las madres, el resto fueron diagnosticadas de DM tipo 2 (4,8%) y otro tipo de diabetes (2,9%). El 9,9% de los pacientes presentaron malformaciones, se observo que aquellos pacientes con malformaciones la HbA1c materna era mayor (6.8 ± 1.8% vs 5,6 ± 1,03%; p=0,000). Al comparar el diagnóstico de DM tipo 1 con respecto al resto se encuentran diferencias estadísticamente significativas en el peso (total y en DS) del neonato (p<0,01), la longitud (total y del DS) del recién nacido (p<0,01), la glucemia capilar (p< 0,01) y la presencia de malformaciones (p< 0,05). CONCLUSIONES: La DM materna se acompaña de un importante número de cesáreas. El mayor riesgo de macrosomía y malformaciones neonatales se encuentra en los hijos de madre con DM1 y en los que tienen mayores concentraciones de HbA1c.
P1d2d3-086 EL POLIMORFISMO COMÚN rs8111699 EN STK11 SE ASOCIA A FACTORES DE RIESGO METABÓLICO EN MUJERES EMBARAZADAS Y A LA PREVALENCIA DE DIABETES MELLITUS GESTACIONAL P. Soriano Rodríguez (1), A. Megía Colet (1), M. Díaz Silva (2), L. Ibáñez Toda (3), J. Bassols Casadevall (4), A. López Bermejo (4) (1) Hospital Joan XXIII. Tarragona, (2) Hospital Sant Joan de Déu. Esplugues Ll., (3) Hospital Sant Joan de Déu. Esplugues Ll., (4) Hospital de Girona Dr. Josep Trueta. Girona Introducción y objetivos: La ruta intracelular STK11-AMPK regula la sensibilidad a la insulina y media el efecto terapéutico de los fármacos antidiabéticos. Nuestro objetivo fue analizar si un polimorfismo de base única (SNP) en el gen STK11 se asocia a parámetros de riesgo metabólico durante el embarazo y, por ende, puede condicionar la prevalencia de diabetes mellitus gestacional (DMG). Diseño y población: Se analizó el SNP rs8111699 (C528G) en STK11 en un estudio transversal de 565 mujeres embarazadas (322 sin DMG y 243 con DMG), que fueron incluidas de forma consecutiva durante las visitas prenatales en los centros de atención primaria de la región de Girona (no DMG) y en el Hospital Juan XXIII de Tarragona (DMG). Se cuantificó la glucemia e insulinemia en ayunas en el segundo trimestre de la gestación. En mujeres con DMG se midió adicionalmente el péptido C basal. La discriminación alélica fue realizada utilizando la tecnología Taqman. Resultados: En mujeres sin DMG, el alelo mutado G del SNP rs8111699 se asoció con una disminución dosis-dependiente de la insulinemia (media ± SEM: 6.7 ± 0.7 vs. 5.6 ± 0.4 vs. 4.8 ± 0.5 mIU/L, respectivamente en sujetos CC, CG y GG p=0.006). En mujeres con DMG, el mismo alelo se asoció a un menor índice de masa corporal (IMC: 31.3 ± 1.2 vs. 29.4 ± 0.5 vs. 28.3 Kg/m2 ± 0.5) y a menor concentración de péptido C basal (2.3 ± 0.1 vs. 1.9 ± 0.1 vs. 1.6 ± 0.1 ng/mL; respectivamente en sujetos CC, CG y GG; p=0.01 para ambos parámetros). Las diferencias encontradas para el péptido C fueron independientes de las encontradas para el IMC. Finalmente, las mujeres con DMG presentaron una menor frecuencia del alelo G en homocigosis (18.1% vs 25.5%; X2=0.03). |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites