
| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):149-154 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.184 |
| Exposición oral. Seleccionados para premios FSEEP |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
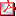 Tabla - PP1/d2-003 Tabla - PP1/d2-003 |
| Tabla - PP1/d2-006 |
|
|
Diabetes PP1/d2-001
MUTACIÓN EN HNF4A ASOCIADA A HIPERINSULINISMO NEONATAL TRANSITORIO Y DIABETES FAMILIAR MONOGÉNICA SUBTIPO HNF4A (MODY1). A. Campos Barros1, C. del Peso Gilsanz2, J.M. Martos Tello3, A. Gómez Núñez4. (1) Hosp. Univ. La Paz; INGEMM, IdiPAZ, UAM y CIBERER (U753), Instituto Carlos III, Madrid. (2) Servicio de Endocrinología, Hospital Reina Sofía, Murcia. (3) Servicio de Endocrinología Pediátrica, Hospital Virgen de la Arrixaca, Murcia. (4) INGEMM, Hosp. Univ. La Paz; IdiPAZ, UAM, Madrid. Introducción: Mutaciones en heterocigosis del gen HNF4A son una causa poco frecuente de diabetes monogénica familiar (<10%). Recientemente se han identificado también mutaciones en HNF4A en casos de hipoglicemia neonatal con respuesta a diazóxido y macrosomía al nacimiento. Objetivos: Establecer el diagnóstico molecular de un caso familiar de sospecha clínica de diabetes monogénica subtipo MODY1 (padre) e hiperinsulinismo neonatal transitorio (2 hijos). Caso clínico: Paciente diagnosticado a los 26 años de diabetes con IMC 26, autoinmunidad negativa (GAD/ ICA), HbA1c 7,5% y microalbuminuria al diagnóstico, con antecedentes familiares de diabetes tipo 2 en padre y madre, inicialmente tratado con insulina y posteriormente con repaglinida. Su historia clínica refleja PN 3,7kg (5,01DE) y TN 50 cm (+2,86DE)(EG: 34 semanas); 1er hijo varón, con hiperinsulinismo neonatal transitorio nacido por cesárea (EG: 37) con PN 3,9 kg (2,25DE) y LN 52 cm (1,85DE) ingresado por hipoglucemia de 21 mg/dl a las 2 horas de vida que presentó buena respuesta a tratamiento con diazóxido, mantenido hasta los 18 meses. Actualmente normoglucémico. 2º hijo varón, con hiperinsulinismo neonatal transitorio nacido por parto eutócico (EG: 38) con PN 3,7 kg (1,36 DE) y LN 52 (1,5 DE) ingresado por episodio de hipoglucemia de 27 mg/dl controlada bien con glucosa (iv) 6 mg/kg/min. Actualmente normoglucémico. Estudios moleculares: Análisis mutacional de las secuencias codificantes transiciones intrón/exón y secuencias reguladoras conocidas de HNF1A y HNF4A del paciente índice y ambos hijos mediante HRM y secuenciación de las variantes detectadas. Análisis de deleciones/ duplicaciones de HNF4A mediante MLPA. Resultados: El estudio molecular detectó la mutación puntual, c.48C>A, p.Tyr16*, en el exón 1D (P2) del transcrito NM_175914.4 de HNF4A en heterocigosis en el índice y ambos hijos. La mutación, introduce un codón de terminación prematuro en el extremo amino terminal, por lo que es predecible que genere una proteína severamente truncada no funcional. Conclusiones: La haploinsuficiencia de HNF4A puede aparecer asociada con un fenotipo variable de diabetes tipo MODY en el adulto e hiperinsulinismo neonatal persistente o transitorio sensible a diazóxido. Se recomienda el consejo genético a familias con mutaciones en heterocigosis de HNF4A para prevenir el riesgo de hiperinsulinismo hipoglucemiante neonatal y macrosomía.
PP1/d2-002
MONITORIZACIÓN CONTINUA DE LA GLUCOSA EN MENORES DE 1500 GRAMOS. J.L. Gómez Llorente, MM Fernández Martínez, J. Momblan de Cabo, I. Alias Hernández, B. Hernández Sierra, A. Bonillo Perales. C.H.Torrecardenas, Almería. Las alteraciones de la homeostasis de la glucosa, la hipo y la hiperglucemia, son los trastornos metabalicos mas prevalentes en el recien nacido de muy bajo peso (<1500 g); ademas han sido relacionados con aumento de la morbilidad, principalmente neurologica. En las Unidades de Neonatologaa la glucemia se monitoriza mediante glucemias capilares, que dan una informacion puntual de los niveles glucémicos. Actualmente existen dispositivos capaces de medir las concentraciones de glucosa de forma continua, lo que permite conocer el perfil glucémico de un paciente durante las 24 horas . Material y métodos: Se realiza la monitorizacion continua de una cohorte de 19 recién nacidos menores de 1500 gramos a partir de las 24 horas de vida y durante los primeros siete días de vida. Se analiza el número y duración de hipoglucemias e hiperglucemias, definiendo hipoglucemia como menor de 40 mg/dl e hiperglucemia mayor de 150 mg/dl. Resultados: La edad gestacional media es de 30,1 semanas (+/-2,71), el peso al nacimiento 1195 gramos (+/- 255). La máxima glucemia media el primer día fue 124,1 mg/dl (+/-30,6), la mínima glucemia media durante el primer día fue 91,39 mg/dl (+/- 29,17), 9 pacientes (47,3%) presentaron hiperglucemias siendo el tiempo medio de 3,67 horas (+/- 7,2). Se detectó hipoglucemias en 3 pacientes 15,78%, el tiempo medio en hipoglucemia fue 1 hora (+/- 3,7). Las cifras de glucemia más bajas se hallaron los días segundo, tercero y cuarto. La correlación entre los datos detectados por el sensor y las glucemias capilares de 11,06 mg/dl (+/- 3,46). Conclusiones: Existe una estrecha correlación entre los valores dados por el sensor de monitorización y las glucemias capilares, por lo que la utilización de este dispositivo para monitorización continua de la glucemia es un método fiable y útil. Destaca la baja frecuencia de hipoglucemias halladas en esta muestra frente a los datos aportados en la bibliografía. La hiperglucemia es el trastorno más prevalentemente hallado, presentándose con una frecuencia similar a la descrita en trabajos anteriores. Gracias a este tipo de seguimiento se pueden detectar alteraciones de la homeostasis de la glucosa que de otra forma pasarían desapercibidas.
Crecimiento PP1/d2-003
CRECIMIENTO PREPUBERAL Y PUBERAL EN 25 MUJERES CON HIPERPLASIA SUPRARRENAL CONGÉNITA. M. Gussinyé Canadell, A. Núñez Mejía, M. Clemente León, M. Albisu Aparicio, A. Campos Martorell, A. Carrascosa Lezcano. Hospital Universitari Vall d’Hebron, Barcelona. Introducción: Estudio longitudinal retrospectivo de crecimiento en 25 mujeres diagnosticadas de hiperplasia suprarrenal congénita (HSC) en el periodo neonatal nacidas entre 1980-1995. Comparación con otra serie nacida entre 1965-1980 (Med Clin Barc 1997;108:87-90) y con el Estudio Español Longitudinal de Crecimiento 2010 (EELC 2010), www.estudiosdecrecimiento.es. Las dosis medias de hidrocortisona (mg/m² sc/ día) fueron 21.83±3.62 en las nacidas 1965-80 y 15.75±2.30 en las nacidas 1980-95. Resultados: En la Tabla 1 se recoge la evolución de la talla; las nacidas entre 1965-80 se compararon con Tanner y las nacidas entre 1980-95 con EELC 2010. En la Tabla 2 se recoge el crecimiento puberal. Según la edad de inicio del brote de crecimiento puberal de las 25 nacidas entre 1980-95, 7 fueron maduradoras muy tempranas, 3 tempranas, 8 intermedias, 4 tardías y 3 muy tardías. Conclusiones: En ambas poblaciones se observa una pérdida importante de talla durante el primer año de vida que se recupera progresivamente pero no de forma total durante el crecimiento prepuberal. En la población nacida 1965-80 esta pérdida se incrementa durante el crecimiento puberal, situación que no sucede en la población nacida 1980-95 que presenta un crecimiento y desarrollo puberal similar a la población del EELC 2010.
Miscelánea PP1/d2-004
¿NOS INTERESA LA ÉTICA? RESULTADOS DE UNA ENCUESTA. G. Martí Aromir1, I. Díez López2, M. Chueca Guindulain3, L. Castro Feijóo4, El. Blarduni Cardón5, M.V. Borrás Pérez6, M.T. Muñoz Calvo7, P. Terradas Mercader8, I. Riaño-Galán9. (1) Hospital Sant Joan de Déu de Manresa/Pediatría, Manresa/Barcelona. (2) Hospital Universitario de Álava. Vitoria. (3) Hospital Virgen del Camino. Pamplona. Navarra. (4) Hospital Clínico Universitario de Santiago de Compostela. Santiago de Compostela. A Coruña. (5) Hospital Zumarraga. Zumarraga. Guipúzcoa. (6) Hospital General de Granollers. Granollers. Barcelona. (7) Hospital Infantil Universitario Niño Jesús. Madrid. (8) Pius Hospital de Valls. Valls. Tarragona. (9) Hospital San Agustín. Avilés. Asturias. Introducción: La creciente complejidad técnica y los cambios en la relación clínica hacen indispensable incorporar la reflexión ética en nuestra práctica clínica, desde un horizonte de excelencia. Objetivos: Conocer la difusión del Grupo de Bioética de la SEEP y sus documentos (en concreto el Código Ético aprobado en 2008) así como los conocimientos y actitudes acerca del respeto a la autonomía y confidencialidad en la práctica clínica. Material y métodos: Encuesta anónima de 17 preguntas con opción múltiple realizada en Google docs, distribuida por correo electrónico a todos los socios de la SEEP con dirección válida (también accesible a través de enlace en la página web de la SEEP). Análisis estadístico descriptivo. Resultados: Respondieron 68 socios (índice de respuesta 35%), con 24,2 años de media de práctica profesional, 58% mujeres. 93% conocen la existencia del Grupo. 28% refieren haber leído el Código Ético y 65% lo habían ojeado. 37% lo valoran útil para la toma de decisiones difíciles y 31% por cuestiones legales. 75% reconocen que en su práctica habitual ayudan al paciente y familia en la toma de decisiones frente al 18% que explican las diversas opciones a la familia dejando que ellos decidan. Un 41% manifiesta haber leído la ley 41/2002. 28% conoce que la mayoría de edad sanitaria es a los 16 años con excepciones. 78% utilizan formularios escritos de consentimiento informado. 72% manifiestan ocultar los datos identificativos de sus pacientes y 38% confiesan que no han cedido nunca su clave de acceso informática. 7% se han comunicado con el Grupo y 40% desconocen cómo consultar. 90% consideran interesante incluir temas de Ética en Cursos y Congresos. La confidencialidad con la historia clínica informatizada, el respeto a la autonomía y la aplicación del consentimiento informado, la investigación clínica y el uso de fármacos fuera de indicación son algunos de los temas de ética propuestos. Conclusiones: Se conoce la existencia del Grupo y se manifiesta interés en su actividad. Se detectan dudas en temas éticos. La ampliación de la formación en aspectos éticos mejorará la atención a nuestros pacientes, en calidad y calidez.
Tiroides
PP1/d2-005
CONCORDANCIA INTEROBSERVADOR EN LA PALPACIÓN DEL TIROIDES EN ESCOLARES DE 6 A 12 AÑOS. A. Muñoz Serrano1, M.P. Falero Gallego2, A. González González3, J.M. Tenías Burillo2, R. Cañete Estrada4. (1) H. Mancha Centro. Servicio Pediatría, Alcázar de San Juan, C. Real. (2) H. Mancha Centro. Alcázar de San Juan. (3) Hospital General Universitario de Ciudad Real. (4) Hospital Universitario Reina Sofía. Córdoba. Introducción: La exploración física, método de elección de detección de bocio en estudios poblacionales por fácil implantación, estandarización de resultados y bajo coste. Esencial demostrar buena concordancia interobservador. Objetivos: Estimar concordancia interobservador en detección de bocio por exploración en población escolar de un área de Salud. Material y métodos: Estudio transversal. Población: 13.896 niños. Muestra: 1134 de 6-12 años. Área de salud: Mancha Centro (Ciudad Real). Centros escolares: 73. Muestreo por conglomerados bietápico. Primera fase: selección 20 colegios. Segunda fase: muestreo aleatorio sistemático, 60 niños de cada centro seleccionado. Exploración enmascarada por dos observadores. Tamaño tiroideo (0, Ia, Ib, II y III). Considerando bocio a partir del grado Ia, inclusive. Concordancia en relación: edad, sexo, índice de masa corporal (IMC), talla, y día de exploración. Medida de concordancia: índice kappa. Resultados: Concordancia en identificación de bocio: Detección bocio: 8,8% observador 1; 9,3% observador 2. Diferencias no estadísticamente significativas (p=0,58). Grado concordancia interobservadores (kappa 0,55; IC95% 0,46-0,64). Concordancia en graduación del bocio: kappa ponderado 0,61; IC95% 0,51-0,71. Cambios en concordancia interobservador en el estudio: estable en todo el estudio (kappas 0,50-0,60) Concordancia interobservadores relacionadas con edad, sexo y variables antropométricas: grado de acuerdo mayor, en niñas y escolares de mayor edad, peso, talla e IMC. Variable más relacionada: talla. Conclusiones: 1. Detección de bocio por palpación en nuestro estudio, concordancia interobservador: óptima e invariable durante todo el periodo de realización. 2. Comparación otras zonas geográficas. Nuestra prevalencia de bocio: 8,8-9,3%.Tres áreas de nuestra comunidad autónoma (1993): 18-24%. 3. Grado concordancia interobservadores en identificación de bocio: en rango medio de lo publicado. Indice estable en todo estudio. Esto puede deberse a que nuestros exploradores poseían amplia experiencia en palpación cervical. 4. Aumento de concordancia con: edad, IMC, peso y, principalmente, con talla; esto puede interpretarse como que la concordancia interobservador es paralela a la misma 5. Índices concordancia más bajos, detectados en subgrupo de niños con características: < 7 años, IMC < 18 kg/m2, peso < 33,2 kg, talla < 135,8 cm. Resultados que permiten compartir opinión: que la exploración cervical no es método diagnóstico fiable de bocio en niños más pequeños.
Genética
PP1/d2-006
SÍNDROME CARDIOFACIOCUTÁNEO: HALLAZGOS CLÍNICOS Y MOLECULARES EN 10 CASOS Y COMPARACIÓN CON 130 PACIENTES CON OTROS TRASTORNOS DE LA VÍA RAS-MAPK. A. Carcavilla Urquí1, S. García Miñáur2, A. Pérez Aytés3, T. Vendrell Bayona4, E. Guillén Navarro5, I. Pinto Fuentes6, J. Sánchez del Pozo7, L. Galbis Martínez8, L. Santomé Collazo8, J.P. López Siguero9, B. Ezquieta Zubicaray8. (1) Hospital Virgen de la Salud. Servicio de Pediatría. Toledo. (2) Instituto de Genética Médica y Molecular. HU La Paz. Madrid. (3) Unidad de Genética Clínica, Hospital La Fe. Valencia. (4) Unidad de Genética Clínica, Hospital Vall d’Hebron. Barcelona. (5) Genética Clínica. Servicio de Pediatría. Hospital Virgen de Arrixaca. Murcia. (6) Servicio de Pediatría. Hospital Severo Ochoa. Leganés, Madrid. (7) Servicio de Pediatría. Hospital Doce de Octubre. Madrid. (8) Laboratorio Diagnóstico Molecular. Servicio de Bioquímica. Instituto de Investigación Sanitaria Gregorio Marañón (IiSGM). Madrid. (9) Servicio de Pediatría. Hospital Carlos Haya. Málaga. Introducción y objetivos: La familia de los síndromes neuro-cardiofaciocutáneos es un grupo de enfermedades con características clínicas compartidas debido a alteraciones en la regulación de la vía de señalización intracelular RAS-MAPK, entre los que se encuentran el síndrome de Noonan y el síndrome cardiofaciocutáneo. Presentamos los datos clínicos y moleculares de 10 pacientes con diagnóstico de síndrome cardiofaciocutáneo, y su comparación con una serie de 130 pacientes con otros síndromes neuro-cardiofacio-cutáneos (19 pacientes con síndrome LEOPARD y 111 con síndrome de Noonan). Pacientes y métodos: Se obtuvieron datos clínicos de pacientes remitidos para estudio genético mediante una base de datos tipo access. Se procedió a amplificación mediante PCR y posterior secuenciación total, o parcial dirigida a los exones más recurrentes, de los genes PTPN11, RAF1, HRAS, KRAS, BRAF, y SOS1. Se obtuvieron datos clínicos de un paciente con estudio positivo para MAP2K1 y otro positivo para BRAF realizados en otro laboratorio. El análisis estadístico se llevó a cabo usando el test exacto de Fisher utilizando el paquete estadístico SPSS 19.0. Resultados: Las características fenotípicas de los pacientes y la mutación identificada se detallan en tabla aparte. Los pacientes con síndrome cardiofaciocutáneo asociaron mayor frecuencia de talla baja, anomalías ectodérmicas, defecto de audición y retraso psicomotor, en comparación con el resto de síndromes neuro-cardiofaciocutáneos (p<0,05). No se encontraron diferencias significativas en sexo, frecuencia y tipo de cardiopatía, criptorquidia, o anomalías torácicas. Los problemas de alimentación fueron más frecuentes (44% frente al 16%) sin llegar a alcanzar significación estadística (p=0,061). En al menos 2 casos, el estudio molecular reorientó el diagnóstico inicial. Conclusiones: El estudio molecular es una herramienta valiosa en el diagnóstico de los síndromes neuro-cardiofaciocutáneos. En nuestra muestra el síndrome cardio-facio-cutáneo se asocia fuertemente a mutaciones en BRAF, anomalías ectodérmicas, defectos de audición, retraso psicomotor y anomalías en la resonancia magnética cerebral. Aunque en general se trata de pacientes con variantes graves, describimos al menos dos pacientes con retraso motor pero desarrollo intelectual dentro de límites normales, lo que ilustra la variabilidad de expresión fenotípica de las mutaciones en BRAF.
Metabolismo y Nutrición
PP1/d2-007
SPRY2 PLACENTARIO: RELACIÓN CON LA DIABETES GESTACIONAL Y EL CRECIMIENTO FETAL. J. Bassols Casadevall1, G. Carreras Badosa1, M. Díaz Silva2, A. Prats Puig1, L. Ibáñez Toda2, A. López Bermejo1, JM. Moreno Navarrete3, JM. Fernández-Real Lemos3. (1) Instituto de Investigación Biomédica de Girona, Girona. (2) Hospital Sant Joan de Déu, Barcelona. (3) CIBERobn. Girona. Introducción: La proteína SPROUTY2 (SPRY2) se ha relacionado con procesos metabólicos y de crecimiento. Estudios recientes sugieren que el SPRY2 puede modular el crecimiento de la placenta. Objetivo: Nuestro objetivo fue estudiar si la expresión de SPRY2 en placenta se asocia a alteraciones del curso normal de la gestación, tales como con la diabetes gestacional (DG) y el hipocrecimiento fetal. Materiales y métodos: Se cuantificó la expressión de SPRY2 y de genes proinflamatorios relacionados con alteraciones de la función de la placenta (TNFα, MMP2 y CD163) en muestras de placenta recogidas al parto en 250 mujeres embarazadas [200 mujeres con embarazos normales, 25 mujeres con DG y 25 mujeres cuyos recién nacidos fueron pequeños para la edad gestacional (PEG)]. Se recogió el valor de la glucosa en la prueba de O’Sullivan y se cuantificaron las concentraciones maternas de péptido C y adiponectina de alto peso molecular (APM) en ayunas en el segundo trimestre de la gestación. Se cuantificó también el peso de la placenta y del recién nacido al parto. Resultados: La expresión de SPRY2 se asoció con la expresión de TNFα, MMP2 y CD163 en placenta. La expresión de SPRY2, MMP2 y CD163 fue mayor en mujeres con DG (p<0.001) y menor en mujeres con niños PEG (p<0.05 a p<0.005). En las mujeres con embarazos normales, la expresión de SPRY2 se asoció a menor adiponectina de APM y mayor glucosa O’Sullivan, péptido C y peso de la placenta (p<0.05 a p<0.0001). Conclusiones: En mujeres embarazadas, la expresión de SPRY2 en placenta se asocia con un fenotipo proinflamatorio, un perfil metabólico desfavorable y mayor crecimiento placentario. Estos resultados sugieren un papel clave de SPRY2 en la función placentaria y por ende en alteraciones del curso normal de la gestación.
Displasias óseas
PP1/d2-008
ANÁLISIS DE MUTACIONES DEL GEN FGFR3 EN PACIENTES CON HIPOCONDROPLASIA SIN PRESENCIA DE LAS MUTACIONES FRECUENTES. S. Benito Sanz1,2, M. Gayo Escribano1, B. Paumard Hernández1, M.P. Bahillo Curieses3, M. Guitart4, J. Sánchez del Pozo5, I. Riaño-Galán6, K.E. Heath1,2. (1) Instituto de Genética Médica y Molecular (INGEMM), IdiPAZ, Hospital Universitario La Paz (HULP), UAM, Madrid. (2) CIBERER, ISCIII. Madrid. (3) Servicio de Pediatría, Hospital Clínico Universitario de Valladolid. (4) Laboratorio de Genética, UDIAT-Centre Diagnòstic, Corporació Parc Taulí, Sabadell. (5) Sección de Endocrinología Infantil, Hospital Universitario Doce de Octubre, Madrid. (6) Servicio de Endocrinología Pediátrica, Hospital Universitario La Paz, Madrid. Introducción: La hipocondroplasia (HCH) es una displasia esquelética, caracterizada principalmente por talla baja desproporcionada debido a un acortamiento rizomélico y lordosis lumbar. Es un trastorno autosómico dominante asociado a mutaciones de ganancia de función en FGFR3. El ~60% de los casos de HCH se explican por una mutación común (p.N540K) y ~2% por las mutaciones: p.N328I, p.I538V y diferentes cambios en los aminoácidos 540 y 650. Mutaciones en otras regiones del gen FGFR3 han sido descritas en una pequeña proporción de pacientes con HCH. Objetivo: Búsqueda de mutaciones en regiones del gen FGFR3 no analizadas de forma rutinaria, en pacientes con HCH sin defecto molecular conocido. Métodos: Estudiamos una cohorte de 61 casos con HCH, en los que se habían descartaron mutaciones frecuentes en FGFR3. Realizamos un análisis mutacional de los exones 1-7,9-11,13 y 15-18 de FGFR3 mediante HRM y secuenciación. La patogenicidad de los cambios observados se determinó mediante estudios de cosegregación genotipo: fenotipo, evaluación de su frecuencia en controles y por herramientas bioinformáticas. Resultados: Identificamos nueve cambios en FGFR3 en 12 pacientes con HCH. Tres son mutaciones patogénicas ya descritas (p.S279C (2), p.L377R, p.R669G), cuatro se descartaron como patogénicas (p.D139A, p.F195L, p.F384L (2), p.P449S (2)) y dos requieren de análisis funcional para determinar su patogenicidad (p.A352V, p.L608V). Conclusiones: 1) Una proporción significativa de nuestra cohorte (13,1%) presenta una mutación patogénica en las regiones alternativas de FGFR3. La mutación p.S279C ha sido observada anteriormente en dos pacientes con acondroplasia, con el tiempo, uno de ellos, ha mejorado su fenotipo, más parecido a una HCH. Sería interesante realizar un seguimiento de dichos pacientes para valorar su evolución fenotípica, esto permitirá dar un mejor consejo genético a futuros casos. 2) La determinación de la patogenicidad de los nuevos cambios en FGFR3 es complicada por la alta frecuencia de variantes no-patogénicas y la implicación de dicho gen en múltiples patologías. 3) Todavía existe una alta frecuencia de pacientes diagnosticados con HCH o fenotipo similar sin diagnóstico molecular. La secuenciación masiva será clave para el estudio completo de FGFR3 y genes candidatos en dichos pacientes. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites