
| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):237-245 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.194 |
| Suprarrenales |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
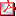 Tabla - P2/d3-150 Tabla - P2/d3-150 |
| Tabla - P2/d3-152 |
|
|
P2/d3-147 SÍNDROME IMAGe. CAUSA INFRECUENTE DE INSUFICIENCIA SUPRARRENAL PRIMARIA N. Pons Fernández, S. León Cariñena, C. de Mingo Alemany, F. Moreno Macián Hospital La Fe, Valencia Introducción: El síndrome IMAGe (retraso del crecimiento intrauterino, displasia metafisaria, hipoplasia adrenal congénita y anomalías genitales) es una causa rara de insuficiencia suprarrenal primaria, de presentación generalmente en el periodo neonatal. Recientemente se han identificado mutaciones en el alelo materno del gen CDKN1C (ganancia de función) como causantes del síndrome. Caso clínico: Niña de 23 meses de edad controlada en Endocrino Infantil por insuficiencia suprarrenal primaria de debut neonatal. Antecedentes personales: embarazo controlado, retraso del crecimiento intrauterino detectado en el segundo trimestre,amniocentesis 46 XX. Cesárea urgente a las 32 semanas por RCIU severo y alteraciones del registro fetal. Antecedentes familiares: primera gestación de padres sanos cosanguíneos (primos hermanos), naturales de Marruecos. No muertes perinatales. Exploración física al nacimiento: Peso 885 g (DE -2,38), T36 cm (DE -4,13), PC 26. Hiperpigmentada, aspecto desnutrido, facies peculiar con frente amplia, desproporción cráneo-facial. Genitales femeninos normales. A los 5 días de vida presenta cuadro de hiponatremia (123 mEq/L), hiperpotasemia (8,4 mEq/L), patrón de pérdida salina en orina (Na/K 6/1 y 9/2), acidosis metabólica e hipoglucemias, que junto a la hiperpigmentación cutánea hace sospechar insuficiencia suprarrenal primaria. El estudio hormonal confirma el diagnóstico: ACTH >1.250 pg/ml, (VN: 9-40) Cortisol no muestra, Aldosterona 36 pg/ml, (VN:42-202) renina plasmática 440 pg/ml (VN:2,5- 62), ARP: muestra insuficiente, 170H progesterona 0,2 ng/ml. Ecografía suprarrenal:normal. Se inicia tratamiento con hidrocortisona, fluorcortisona y suplementos de Sodio. Asocia una nefrocalcinosis en patrón microlitiasis, Ca sangre: 8,9-11 mg/dl, Ca/Crorina 0,52, atribuida al tratamiento diurético y una colestasis con hepatitis de células gigantes por CMV. A lo largo de su seguimiento destaca un retraso del crecimiento postnatal, presentando a los 12 meses: Talla a -3,7 DE, Peso -4,9 DE, rasgos físicos característicos: frente olímpica, raíz nasal deprimida, micrognatia, parecido a síndrome de Silver Russell. Exploraciones realizadas: serie ósea: sin hallazgos patológicos, eco pélvica no se visualizan ovarios ni útero, potenciales evocados normales. Pendiente el estudio genético. Conclusiones: El síndrome IMAGe es un trastorno con amplio espectro fenotípico. La ausencia displasia metafisaria en nuestra paciente no excluye el diagnóstico. La identificación genética de una insuficiencia suprarrenal es fundamental para realizar un diagnóstico, tratamiento y consejo genéticos adecuados.
P2/d3-148
CARACTERÍSTICAS GENÉTICAS Y FENOTÍPICAS DE HSC FORMA NO CLÁSICA: REVISIÓN DE NUESTRA CASUÍSTICA S. Berrade Zubiri(1), J. Alvarez García(1), A. Sagastibelza Zabaleta(1), M. Chueca Guindulain(1), B. Ezquieta Zubicaray(2), M. Oyarzabal Irigoyen(1) (1) Complejo Hospitalario de Navarra, Pamplona; (2) Hospital Gregorio Marañón. Madrid Introducción: La hiperplasia suprarrenal congénita (HSC) en su forma no clásica es una entidad frecuente (0.3%), con amplia variabilidad clínica y genética. Objetivos: Descripción de los casos de hiperplasia suprarrenal congénita no clásica diagnosticados en la Unidad de Endocrinología Pediátrica de nuestro hospital entre 1980-2012. Método: Se ha solicitado estudio genético por sospecha de HSC a un total de 39 niños/as, con confirmación diagnóstica de HSC tardía en 21 casos, portadores de mutación 13 casos y pendientes de resultados 4 pacientes. Revisamos los 21 casos de HSC tardía: edad, clínica y analítica al diagnóstico, genética molecular, evolución, desarrollo de complicaciones y talla final. Resultados: 21 pacientes (5 V/16 M), con edad media al diagnóstico 7,7 ±1,5 años (4,9-10,6) y motivo de consulta: pubarquia precoz (20 casos) y aceleración de edad ósea (1 caso); presentaban talla: +0,7 SDS, peso: +2,8 SDS e IMC: +0,47 SDS, y adelanto en la edad ósea de 1,3±1,1 años respecto a la edad cronológica. 17OHProgesterona basal 18,2±12,1ng/ml y post- ACTH:59,6±24.4 ng/ml. El análisis genético muestra 9 casos con alteración del gen en homocigosis (todos ellos Val281/Val281), 11 en heterocigosis compuesta (10 mutaciones severas) y un caso no caracterizada. Un 52 % recibieron tratamiento con hidrocortisona (7H/3M); el 12 % de las mujeres requirió análogos GnRH por pubertad precoz, 18% antiandrógenos por hirsutismo y el 31 % ACO por trastornos menstruales. Del total, 16 pacientes (3V/13M) han finalizado crecimiento, no objetivando diferencias significativas entre talla final y talla genética. Conclusiones: • La HSC no clásica, ha tenido como principal y casi único motivo de consulta la pubarquia precoz. Las principales complicaciones se deben al hiperandrogenismo, sin afectación reseñable en la talla final. • El estudio genético molecular ha confirmado el diagnóstico en un 95% de los casos. La mutación más frecuente ha sido Val281, y 10 pacientes son portadores de mutación severa.
P2/d3-149
CARACTERÍSTICAS GENÉTICAS Y FENOTÍPICAS DE HSC FORMA CLÁSICA: REVISIÓN DE NUESTRA CASUÍSTICA S. Berrade Zubiri(1), J. Alvarez García(1), M. Chueca Guindulain(1), A. Sola Mateos(1), B. Ezquieta Zubicaray(2), M. Oyarzabal Irigoyen(1) (1) Complejo Hospitalario de Navarra, Pamplona; (2) Hospital Gregorio Marañón. Madrid Introducción: La hiperplasia suprarrenal congénita (HSC) en-globa un grupo de enfermedades, de herencia autosómica recesiva, causadas por la deficiencia de alguna de las enzimas responsables de la síntesis de cortisol. En el 95% de los casos la alteración enzimática implicada es la 21 hidroxilasa. Objetivos: Descripción de los casos de HSC forma clásica diagnosticados en la Unidad de Endocrinología Pediátrica de nuestro hospital entre 1977-2012. Método: Se han diagnosticado un total de 10 pacientes (4 varones/6 mujeres), el 80% forma pérdida salina y el 20% forma virilizante simple. Se revisan edad, clínica y analítica al diagnóstico, genética molecular, evolución, complicaciones y talla final. Resultados: Edad media al diagnóstico de 14 días (rango:1-43), motivos del mismo: ambigüedad sexual (54%), crisis adrenal (36%) e hiperpigmentación genital (9%), 17OH Progesterona basal: 337,6±274,8 ng/ml (110- 690). El análisis genético muestra en 4 casos mutaciones severas en homocigosis y 6 en heterocigosis compuesta, siendo la más frecuente Gln318Stop. El estudio de los familiares ha detectado 6 hermanos portadores de mutación severa. De las 6 niñas, presentaban virilización de Prader: grado II (1 caso), grado III (1 caso), grado IV (3 casos) y grado V (1 caso). La corrección quirúrgica se realizó en la primera infancia, a una edad media de 23 meses (6-36), y un caso ha precisado reintervención en edad adulta. 7 pacientes (3V/4M) han finalizado crecimiento, con talla final similar a talla genética. Complicaciones: 1 paciente precisó ingreso por crisis adrenal durante un proceso intercurrente por mal ajuste de la medicación, 1 niña desarrolló pubertad precoz que fue tratada con análogos GnRH, 2 casos (edad adulta) precisaron antiandrógenos por hirsutismo, 1 varón, con mal seguimiento del tratamiento sustitutivo, desarrolló tumor testicular bilateral y 1 mujer ha tenido trastornos psiquiátricos (anorexia y depresión). Conclusiones: • La evolución clínica de estos pacientes ha sido, en general, satisfactoria, con escasas complicaciones por la enfermedad y buena talla final. • El análisis molecular ha permitido la caracterización de mutaciones severas del gen de la 21-hidroxilasa, siendo la más frecuente Gln318Stop. • 6 hermanos de pacientes han resultado portadores de mutaciones severas y han recibido el consiguiente consejo genét.
P2/d3-150
TUMORES ADRENOCORTICALES EN LA EDAD PEDIÁTRICA N. Pons Fernández, C. De Mingo Alemany, S. León Cariñena, F. Moreno Macián Hospital la Fe, Valencia; Hospital la Fe, Valencia Introducción: Los tumores adrenocorticales constituyen menos del 0.2 % de las neoplasias en la edad pediátrica. El 60 % de los casos se produce en menores de 4 años y la mayoría son funcionantes. Se deben sospechar ante clínica cushingoide o de hiperandrogenismo. Material y métodos: Estudio de cohortes retrospectivo. Análisis descriptivo. Resultados: Muestra 5 pacientes: 3 mujeres, 2 varones. Edad media al diagnóstico 4,29 años (DS 5,93). La forma de presentación: síndrome de Cushing en 3 pacientes (2 adenomas y 1 carcinoma), y virilización en 2 (1 adenoma y 1 carcinoma). No se halló patrón de secreción mixta o de hiperaldosteronismo. En la Tabla 1 se muestran los datos de laboratorio. De entre los pacientes con síndrome de Cushing: en uno se observó pérdida del ritmo nictameral, en dos elevación del cortisol plasmático, y en todos ellos elevación de la cortisoluria, media 311,57 microg/24h (DS 174,39). La elevación de testosterona se detectó en una niña que desarrolló rápida virilización. El tratamiento quirúrgico se realizó en 4 niños por laparotomía y en uno por laparoscopia. Los adenomas presentaron un peso medio de 50 gr e histología Weiss menor de 3 puntos. De los carcinomas uno tuvo un peso mayor de 200 gr, y el otro fue un tumor grande y fragmentado, ambos tenían una puntuación de Weiss mayor de 3 puntos. Las pruebas de imagen de los pacientes con carcinoma demostraron en uno una masa con diámetro máximo 8,5 cm, y en el otro una masa con diámetro máximo 5,5 cm. El primero presentó metástasis a los pocos meses, precisó quimioterapia y metastectomía pulmonar. Como reacción secundaria al mitotane desarrolló hipotiroidismo y al año del diagnóstico desarrolló una pubertad precoz central, sobrevive 5 años después. El segundo caso falleció, era una niña de un mes de vida con carcinoma secretor de cortisol. Conclusiones: La diferenciación entre adenoma y carcinoma es compleja, pero es fundamental para el pronóstico y el seguimiento. El diámetro máximo de la masa adrenal es predictivo de malignidad. La resección tumoral completa es el indicador pronóstico individual más importante. El tamaño tumoral también tiene valor pronóstico.
P2/d3-151
EVOLUCIÓN ATÍPICA DE UN CASO DE INSUFICIENCIA SUPRARRENAL PRIMARIA F.J. Rodríguez Contreras(1), L. Salamanca Fresno(1), L. Gutiérrez Pascual(1), A.C. Barreda Bonis(1), R. Álvarez Doforno(2), I. González Casado(1) (1) Hospital Universitario La Paz / Servicio de Endocrinología Pediátrica, Madrid; (2) Hospital Universitario La Paz / Servicio de Inmunología. Madrid Introducción: La etiología autoinmune supone la causa más frecuente de insuficiencia suprarrenal primaria (enfermedad de Addison), seguida por la administración de fármacos, infecciones y la enfermedad metastática. Entre las menos frecuentes destaca la adrenoleucodistrofia por acúmulo de ácidos grasos de cadena muy larga en diversos tejidos. Se presenta el caso de una paciente con diagnóstico de adrenalitis autoinmune con hallazgos en la neuroimagen compatibles con leucodistrofia. Resumen: Mujer caucásica de 12 años que acude a Urgencias por cuadro de vómitos de 24 horas de evolución. Como antecedentes destaca una tiroiditis linfocitaria en tratamiento sustitutivo desde hace 13 meses. La familia refiere cuadro de astenia, anorexia, pérdida de peso y avidez por la sal desde hace 6 meses. A la exploración destaca hiperpigmentación, decaimiento e hipotensión arterial. En analítica sanguínea se objetiva glucemia de 66 mg/dL, natremia de 122 mmol/L y discreta acidosis metabólica, patrones que se repiten en los controles analíticos. El estudio analítico confirma ACTH > 1.250 pg/mL con cortisol basal < 1 mcg/ dL, actividad de renina plasmática de 16,4 ng/ mL/h, aldosterona < 25 pg/mL y anticuerpos antiadrenales positivos. Se inicia tratamiento sustitutivo con hidrocortisona y 9 alfa Fluorhidrocortisona. Ante la posibilidad de un síndrome poliglandular autoinmune se efectúa estudio de autoinmunidad objetivando anticuerpos antihipófisis a título muy positivo. Se realiza RNM del área selar/supraselar que no objetiva engrosamiento infundibular, sino marcada afectación de sustancia blanca subcortical con adelgazamiento significativo del cuerpo calloso, datos muy sugestivos de leucodistrofia. Ante estos datos se solicita estudio de ácidos grasos de cadena muy larga. La paciente se mantiene asintomática neurológicamente. Conclusiones: Las mujeres portadoras heterozigotas de adrenoleucodistrofia pueden mostrar anomalías neurológicas leves ó lentamente progresivas, y raramente desarrollan insuficiencia suprarrenal. No en todos los casos se observan anomalías en la RNM aunque en muchas ocasiones la sustancia blanca subcortical puede afectarse tardíamente. La ausencia de anticuerpos antiadrenales suele ser constante. Llama la atención en nuestra paciente que a pesar de la sospecha de insuficiencia suprarrenal de origen autoinmune, las imágenes de la RNM puedan ser compatibles con leucodistrofia.
P2/d3-152
NUEVA MUTACIÓN PATOGÉNICA EN EL SÍNDROME DE RESISTENCIA A LOS GLUCOCORTICOIDES (RGC) G. Grau Bolado(1), G. Pérez de Nanclares Leal(1), A. Vela de Sojo(1), A. Rodrígues Estévez(1), I. Rica Etxebarria(1), L. Castaño González(1), M.R. Saracho Arbaiza(2) (1) Hospital Universitario de Cruces.UPV/EHU.CIBERER., Bizkaia; (2) Pediatra centro de salud de Llodio. Álava El síndrome de resistencia a los glucocorticoides familiar es una patología poco prevalente ocasionada por mutaciones en el gen del receptor de los glucocorticoides (NR3C1) heredadas de forma autosómica recesiva o dominante. Se caracteriza por una resistencia periférica a la acción del cortisol con el subsiguiente incremento de ACTH y por tanto de glucocorticoides, mineralocorticoides y andrógenos suprarrenales. Caso clínico: Niña de 12 años y 7 meses remitida por hipercortisolismo (cortisolemia 37,4 µg/dl). Su pediatra realizó un estudio hormonal basal ante estría clínicamente irrelevante en un muslo. Menarquia a los 11 años y menstruaciones regulares. No refería clínica asociada, enfermedades intercurrentes, ni ingesta de medicaciones. A la exploración: peso en p75 con talla en p90-97, TA normal, hirsutismo leve (7- 8), una estría en muslo derecho. y desarrollo puberal grado IV. Resto de exploración normal. Pruebas complementarias: • Se confirma hipercortisolismo con ACTH elevada y con ritmo circadiano mantenido. - 1ª determinación: Cortisolemia 30,8 µg/dl. Cortisoluria 24 horas 417 µg. - ACTH 108 pg/ml. - 2ª determinación: Cortisolemia 38,1 µg/dl. Cortisoluria 24 horas 300 µg. - ACTH 121 pg/ml. - Cortisol en saliva 8:00 horas 3,5µg/dl. - Cortisol en saliva 23:00 horas 0,46µg/dl. DHEAS 1150 ng/ml. Testosterona 0,8 ng/ml.17 hidroxiprogesterona 1,6 ng/ml. Renina 0,8 ng/ml/hora. Aldosterona 210 pg/ml. • RMN sin signos sugestivos de alteración hipofisaria. • Pruebas de supresión con Dexametasona: •Ausencia de repercusión clínica del hipercortisolismo. Analítica general: glucemia, iones y lípidograma sin alteraciones. DMO: Columna -1,1 D.S .Cuello femoral normal. • Estudio familiar (ante la sospecha de RGC). Supresión corta DXM: padre: 0,5 µg/dl; madre: 3,7 µg/dl (cortisoluria:62 µg/24h) Estudios genéticos: Se analizó mediante PCR y secuenciación directa el gen NR3C1. Se encontró el cambio en heterozigosis c.1429C>T; p.Arg477Cys en la paciente y en su madre. Esta mutación no ha sido descrita previamente, pero los software de predicción (Polyphen2, SIFT, Mutation T@sting…) la describen como patogénica.
P2/d3-153
LACTANTE CON HIPONATREMIA TRANSITORIA E HIPERALDOSTERONISMO M.J. Pérez Ortega, J.L. Gomez Llorente, M.M. Oña Aguilera, J. Momblan de Cabo, M. Sanchez Forte, A. Bonillo Perales C.H. Torrecárdenas. Servicio de Pediatría, Almería Introducción: El pseudohipoaldosteronismo renal tipo 1 (PHA1) constituye una causa infrecuente de pérdida salina en el lactante y se debe a una falta de respuesta del túbulo renal a la acción de la aldosterona. Se han detectado casos familiares (AD) y casos esporádicos y en ambos se han identificado mutaciones heterocigotas del gen NR3C2. Caso clínico: Neonato mujer de 17 días de vida que ingresa por pérdida de peso, rechazo de la alimentación y leve hiponatremia (Na 130,8 mE/L). Antecedente: abuela materna con enfermedad de Addison. Parto eutócico, 36+5 semanas de EG y peso al nacimiento 2.650 gramos. Ingreso a las 24 horas de vida por hipoglucemia sintomática (<20 mg/dl) y segundo ingreso a los 8 días de vida por escasa ingesta y pérdida de peso del 10%. En analíticas seriadas presenta hiponatremia (130,8 mEq/l) e hiperpotasemia (6 mEq/l), elevación aldosterona (1.435 pg/ ml) y ARP (41 ng/ml/h) siendo remitida a nuestro hospital. Exploración: aspecto desnutrido (peso 2.790 gr P4), ictericia, hipotonía generalizada y llanto débil. Durante su ingreso presentó hiponatremia (129 mEq/l) e hiperpotasemia (6,5 mEq/l). Resto pruebas complementarias normales (iones en orina, EFNa, ecografía renal, electrolitos en sudor, 17 OHPG, ACTH, cortisol y hormonas tiroideas). Inicia tratamiento con aportes orales de sodio con mejoría clínica y normalizándose cifras de sodio y potasio. En seguimiento en consulta, a pesar de tratamiento y normalización de iones presenta aldosterona de 9.424 pg/ml y ARP 103 ng/ml/h. Por la buena evolución se realiza descenso de los aportes extras de sodio, quedando sin tratamiento al mes del inicio del cuadro. A los 3 meses presenta normalización de sodio y potasio con cifras elevadas de aldosterona (6.952 pg/ml) y normalización ARP. Ante la sospecha de PHA1, se solicita estudio genético del gen NR3C2, detectándose mutación tipo missense c.2492 C>S (p.P831R), no descrita previamente en la bibliografía. Comentarios: a) Solo una pequeña proporción de heterocigotos desarrolla un cuadro clínico de pérdida salina y muchos portadores permanecen asintomáticos pero con elevación de la aldosterona en plasma, lo que hace que sea infradiagnosticada. b) La descripción de una nueva mutación posiblemente patogénica no descrita previamente.
P2/d3-154
ESTUDIO DE LA VARIABILIDAD CLÍNICA EN LOS TRASTORNOS SUPRARRENALES PRIMARIOS, ¿ES ÚTIL PARA EL DIAGNÓSTICO? A. Barasoain Millán, O. Patiño Hernández, C. Notario Muñoz, C. Bezanilla Lopez Hospital Universitario Fundación Alcorcón, Madrid Introduccion: La aparición de signos de activación suprarrenal a edades precoces genera preocupación y frecuentes consultas hospitalarias. La existencia de un espectro clínico diferencial que oriente hacia una etiología orgánica o funcional podría ser de utilidad en la búsqueda de valoración médica especializada y en la solicitud de determinaciones bioquímicas hormonales específicas, reduciendo también el gasto sanitario. Objetivo: Establecer y comparar las características clínicas al diagnóstico de los trastornos suprarrenales más frecuentes en la infancia: Hiperplasia suprarrenal congénita no clásica (HSC NC) e Hiperandrogenismo suprarrenal funcional (HSF) , en relación con la población no afecta. Material y métodos: Se recogieron retrospectivamente variables clínicas y demográficas al diagnóstico en pacientes en edad pediátrica sometidos al test de ACTH desde los años 2002-2012 en un hospital secundario. Se comparó el espectro clínico con los resultados de 17OH-Progesterona tras estímulo. Resultados: De los 125 pacientes sometidos al test y con datos clínicos completos, el 96% fueron mujeres. Se catalogaron de HSC NC el 6,4% de los pacientes y un 8,8% de HSF. El 13,7% presentaron prematuridad y un 2% fueron pequeños para la edad gestacional de manera invariable. La edad media al inicio de los síntomas fue 6,7 años (DE0,18) siendo la pubarquia prematura el principal motivo de consulta (65%) con un adelanto óseo de +1,5 años respecto a EC sin cambios entre grupos. Se observó una proporción mayor aunque no significativa de pacientes en estadio P4 (25% vs 8,5%) y Axilarquia +++ (18 vs 5%) en el grupo HSC NC respecto a los sanos. Los pacientes afectos de HSF presentaron valores intermedios en dichas variables. No se observaron diferencias (p>0,05) por grupos en la prevalencia de obesidad (IMC>2DE), acné, hirsutismo, olor apocrino, o edad de inicio de la pubertad. Conclusiones: En nuestro estudio, no se ha logrado identificar variables clínicas que permitan catalogar los diferentes trastornos suprarrenales. El diagnóstico precoz, la limitaciones del diseño del estudio y sobre todo el escaso tamaño muestral de uno de los grupos limita la asociación estadística que se intuye en algunos rasgos clínicos. Estudios más amplios podrán determinar y ponderar el peso de estos hallazgos.
P2/d3-155
HIPONATREMIA SEVERA Y FALLO DE MEDRO I. Mulero Collantes, B. Salamanca Zarzuela, C. Alcalde Martín, A.M. Vegas Álvarez, R. Izquierdo Caballero, J.C. Hernando Mayor Hospital Universitario Rio Hortega. Valladolid Introducción: El hipoaldosteronismo hiperreninémico familiar (HHF) es una rara entidad congénita (1/100.000 recién nacidos vivos) por mutación del gen CYP11B2 (cr.8), que origina un déficit de aldosintetasa, la cual presenta dos actividades enzimáticas, existiendo dos formas clínicas de enfermedad: tipo I o déficit de CMO-I (paso de corticosterona a 18-OH-corticosterona) y tipo II o déficit de CMO-II (18-OH-corticosterona a aldosterona). Se manifiesta precozmente por deshidratación, pérdida de peso o fiebre intermitente. Caso clínico: Neonato de 18 días de vida que ingresa por escasa ganancia ponderal. Antecedentes familiares y personales: no consanguinidad ni enfermedades reseñables. Embarazo, parto, periodo neonatal y somatometría al nacimiento normales. Al ingreso destacamos: escasa ganancia ponderal, leve palidez sin hiperpigmentación y presencia de genitales masculinos normales. TA 64/47 mmHg, FC 174 lpm. Analíticamente objetivamos: Na 117 mmol/L, K 7,3 mmol/L, Cl 84 mmol/L. Hemograma, gasometría y resto de bioquímica normales incluyendo glucemia. Urocultivo: infección urinaria (ITU) por E.coli. No corrigiéndose el trastorno hidroeléctrolítico tras antibioterapia, descartamos hipoaldosteronismo transitorio secundario a ITU. Se completa estudio: hormonas tiroideas normales, cribado neonatal normal, 17-OH Progesterona y cortisol basales, testosterona y delta-4-androstendiona normales para la edad. 18-OH Corticosterona y corticosterona elevadas. Aldosterona muy disminuida (37 pg/ml, VN de 4-30 días: 746±273), ARP muy elevada (>18,1 ng/ml/h, VN de 4-30 días: 9,5±6,6) y 18-OH Corticosterona/aldosterona muy elevado. RMN cerebral y abdominal normales. Estudio gen CYP11B2: dos mutaciones patogénicas en heterocigosis, confirmando el diagnóstico de déficit de CMO II (cambio de A>G en posición c.594 originando el cambio p.Glu198Asp y T>C en posición c.1157 que da lugar a p.Val386Ala). Pautado tratamiento sustitutivo con fluodrocortisona oral (máximo 250 mcg/dia) e hidrocortisona (15 mg/ m2/día) hasta estabilización inicial, suplementos de ClNa (hasta 7 mEq/l) y tratamiento de la hiperpotasemia. Buena ganancia ponderal y normalización iónica, disminuyendo paulatinamente fluodrocortisona y precisando propranolol. Conclusión: Ante un trastorno iónico neonatal descartaremos por su gravedad la hiperplasia suprarrenal congénita. El HHF es infrecuente y puede presentarse como deshidratación con pérdida salina o fallo de medro. El tratamiento es sustitutivo (iónico y hormonal) con buen pronóstico, corrigiéndose parcialmente el déficit salino con la edad.
P2/d3-156
UNA APARENTE HOMOZIGOSIS DEL ALELO LEVE Val281Leu PUDO ENMASCARAR UN CONSEJO GENÉTICO ADECUADO PARA DEFICIENCIA DE 21 HIDROXILASA C. Bezanilla Lopez(1), B. Ezquieta Zubicaray(2) (1) Hospital Universitario Fundación Alcorcón, Madrid; (2) Hospital Universitario Gregorio Marañón, Madrid La hiperplasia suprarrenal congénita (21OHD) es una enfermedad autosómica recesiva, severa, no infrecuente y amplio espectro clínico. La caracterización molecular ha permitido establecer estrecha correlación genotipo-fenotipo permitiendo predecir el comportamiento clínico, a partir del genotipo. Así, homocigotos o heterocigotos compuestos de mutación leve o leve/severa dan lugar a formas leves o no clásicas y los homocigotos o heterocigotos compuestos de mutaciones graves dan lugar a formas severas o clásicas. Una de alelos leves más prevalentes y clásicamente “leve” es la mutación puntual p.Val282Leu (antes Val281Leu), aunque en ocasiones se había encontrado en pacientes con forma pierde sal en heterozigosis compuesta con mutaciones graves, lo que había llevado a interpretar todos los alelos p.Val282Leu como potencialmente severos. En 2010 se describió su asociación a una alteración intrónica (c292+5G>A) en estos casos graves. Aunque el aleo c.292+5G>A;p.Val282Leu es infrecuente en la población podría encontrarse en algunos paciente con formas no clásicas. Presentamos el caso de dos hermanas con forma NC en las que un segundo análisis genético detectó esta variante modificando la necesidad de consejo genético. Niña de 7 años vista en 2002 por pubarquia de un año de evolución, edad ósea no adelantada y 17- OH progesterona basal > 20 ng/ml. Ante la sospecha de HSC se solicita estudio genético en 2009 que confirma la presencia de mutación p.Val282Leu en homozigosis. Entre los familiares destacaba la hermana de 17 años con acné moderado e hirsutismo ya estudiado en otro centro. Se revisó la historia y al hallarse datos analíticos compatibles 21OHD se solicitó en 2010 estudio genético que mostró la presencia no sólo de p.Val282Leu en homozigosis sino también la mutación severa (c292+5G>A) en heterozigosis (c.292+5G>A;p.Val282Leu+p.Val282Leu). Ante este hallazgo se analizó la región intrónica de interés en la muestra de la primera paciente confirmándose que también presentaba el genotipo c.292+5G>A;p.Val282Leu+p.Val282Leu lo que modificó la necesidad futura de un adecuado consejo genético preconcepcional. Dada la importancia del consejo genético derivado, esta variante, aunque infrecuente, es actualmente analizada en el cribado básico practicado.
P2/d3-157
ADRENALECTOMÍA BILATERAL EN SIETE MUJERES CON TALLA ADULTA Y UNA PACIENTE PREPUBERAL CON HIPERPLASIA SUPRARRENAL CONGÉNITA FORMA CLÁSICA DE DIFÍCIL CONTROL M. Clemente León, M. Gussinyer Canadell, A. Nuñez Mejias, D. Yeste Fernández, M. Asensio Llorente, A. Carrascosa Lezcano Hospital Materno-infantil Vall d’Hebron, Barcelona Objetivos: Evaluar los resultados de la adrenalectomía bilateral en mujeres afectas de hiperplasia suprarrenal congénita (HSC) que no se pueden controlar con tratamiento convencional. Pacientes y métodos: Ocho mujeres con HSC clásica con pérdida salina por déficit de 21-hidroxilasa. Se practicó adrenalectomía bilateral por laparoscopia a la edad de 17.3 (8.5-27.6) años. Indicaciones de la adrenalectomía: manifestaciones de hiperandrogenismo con hidrocortisona 15-19 mg/m2/día. Siete presentaban talla adulta, amenorrea primaria con hidrocortisona y efectos secundarios importantes con dexametasona. La paciente prepuberal presentaba aceleración de la edad ósea (+4 años). Previo a la cirugía se realizó en seis pacientes gammagrafia con I¹³¹colesterol para descartar tejido ectópico adrenal en ovario. Tratamiento postadrenalectomía: hidrocortisona 12- 17 mg/m2/día y fluorhidrocortisona 0,1 mg/día. Resultados: Beneficios: en todas desapareció el hiperandrogenismo. Las pacientes postpuberales presentaron reglas regulares a los 2-3 meses. La paciente prepuberal inició botón mamario a los 3 meses de la intervención, realizó brote de crecimiento puberal de 13 cm y alcanzó una talla final de 152 cm. Complicaciones: ninguna paciente presentó complicaciones relacionadas con la cirugía. Seis pacientes presentaron, 2-3 meses tras la cirugía, episodios de astenia, anorexia, vómitos e hipotensión con natremias 133-138 mEq/L, caliemias normales y valores de ACTH muy elevados (>400 pg/ml) a pesar del tratamiento con hidrocortisona a dosis de 20 mg/m2/día y posteriormente con prednisona. La clínica mejoró parcialmente con el tratamiento con etilefrina, en tres de ellas desapareció tras 36-48 meses y en una persiste tras 6 años de la cirugía. En seis se realizaron RM cerebrales seriadas por niveles elevados de ACTH, 2 presentaron adenohipófisis aumentada de tamaño sin imágenes sugestivas de adenoma y en 4 fueron normales. Situación actual: Años tras la cirugía: 6,3 (2-8,5). Analítica: ACTH 231 (16-1.250) pg/mL, 17-hidroprogesterona 1,4 (0,7-3) ng/mL, testosterona 25(<10- 64) ng/dL. Tratamiento: en todas prednisona 5-10 mg/día y en 6 etilefrina 10-30 mg/día. Conclusión: La adrenalectomia bilateral en mujeres con HSC de difícil control corrige el hiperandrogenismo y reinstaura la función gonadal normal. Sin embargo condiciona la aparición de un cuadro de insuficiencia suprarrenal difícil de controlar con el tratamiento con glucocorticoides y mineralcorticoides, transitorio en la mayoría de pacientes.
P2/d3-158
FORMAS NO CLÁSICAS DE LA HIPERPLASIA SUPRARRENAL CONGÉNITA. REVISIÓN DE LOS CASOS DIAGNOSTICADOS EN UNA CONSULTA DE ENDOCRINOLOGÍA PEDIÁTRICA J. Dorca Vila, G. Martí Aromir, L. Bilbao Gassó, E. Botifoll García, S. Burgaya Subirana, M. Garriga Badía Hospital Sant Joan de Déu de Manresa, Barcelona Introducción: La hiperplasia suprarrenal congénita tardía o no clásica (HSC-NC) es una entidad monogénica recesiva frecuente en nuestra población. La existencia de hiperandrogenismo en diferentes etapas de la vida con manifestación clínica variable nos guía hacia el diagnóstico genético. Su detección ayuda a un buen seguimiento, valorar la necesidad de tratamiento, estudios complementarios y orientar su etapa adulta. Objetivo: Valorar los datos clínicos, bioquímicos y la base molecular de los pacientes afectos de HSC tardía controlados en Consultas Externas de Endocrinología Pediátrica de nuestro Hospital. Método: Análisis retrospectivo de los datos clínicos, bioquímicos, base molecular de 7 pacientes diagnosticadas de HSC tardía. Así como su seguimiento y tratamiento. Resultados: 7 pacientes de sexo femenino (edad media 9,8 años, Tanner 2 (n=3), Tanner 3 (n=1), Tanner 5 (n=3). Motivos de consulta; adrenarquia adelantada (n=4), pubertad adelantada (n=2), reglas irregulares (n= 2), hirsutismo (n=1), acné (n=2), sobrepeso (n=2). Datos radiológicos y bioquímicos; edad ósea con un décalage de +2,5 años en las 7 pacientes. Valor medio de 17 hidroxiprogesterona basal (17OHP) 1,78 ng/mL (rango 0,43 ng/mL-6,78 ng/mL) y a los 60 minutos del test de Synacthen 250 µg ACTH) de 11,22 ng/mL (rango 4-48.25 ng/mL). Diagnóstico genético; V281L (n=4, 1 en homozigosis y 3 en heterozigosis), P453S en homozigosis (n=1), P453S en heterozigosis (n=1), intrón 2 G656 en heterozigosis (n=1). Tratamiento;se valoró en 2 pacientes Prednisona 2,5mg/12h asociada a Flutamida y anovulatorio (n=1) y Dexametasona 0,25mg/ día (n=1). Evolución: en los casos tratados mejoría del hirsutismo, reglas regulares y acné. Conclusión: El signo clínico guía en nuestras 7 pacientes fue principalmente la adrenarquia adelantada, siempre acompañada de una edad ósea acelerada. La 17-OH-Progesterona basal no nos orientó al diagnóstico. En test de ACTH fue significativa la elevación de la 17OHP en todos los casos, mayor para los 2 casos de mutación en homozigosis. En nuestra serie se demuestra la prevalencia de la mutación V281L. La actitud tomada para iniciar el tratamiento se ha basado en criterios clínicos. La evolución ha sido favorable en ambos casos y el resto realizan actualmente seguimiento en nuestro Servicio.
P2/d3-159
DÉFICIT PARCIAL DE 3β HIDROXIESTEROIDE DESHIDROGENASA DETECTADO EN SCREENING NEONATAL DE 21 HIDROXILASA. M.P. Bahíllo Curieses(1), I. Alía Arroyo(1), L. Loidi Fernández de Trocóniz(2), F. Hermoso López(1), A. del Cañizo López(3), MJ. Martínez Sopena(1) (1) Hospital Clínico Universitario Valladolid. Servicio Pediatría. Endocrinología Pediátrica, Valladolid; (2) Unidad de Medicina Molecular. Fundación Pública Galega de Medicina Xenómica. Santiago de Compostela; (3) Hospital Clínico Universitario Valladolid. Servicio de Cirugía Pediátrica Introducción: Las anomalías de diferenciación sexual (ADS) 46 XY, son entidades poco frecuentes y de difícil diagnóstico. Un pequeño porcentaje de ellas son secundarias a defectos en la síntesis de testosterona. Presentamos un caso de ADS 46 XY por déficit de 3βhidroxiesteroide deshidrogenasa tipo 2 (3βHSD2) detectado en el screening neonatal de 21 hidroxilasa (21OH). Caso clínico: Niña de 25 días con cifras de 17 OHP 40 ng/ml en screening neonatal de 21OH. Clínicamente asintomática. Antecedentes familiares: 1º/1. Padres jóvenes no consanguíneos. Antecedentes personales: Embarazo, parto y periodo neonatal normales. Asignación de sexo femenino. Exploración física: Leve hipertrofia de clítoris, seno urogenital (Prader II), resto normal. Exploraciones complementarias: Cariotipo 46 XY. Analítica sanguínea normal. 17 OHP 30.3 ng/ml (2,3-9,84), androstenediona 2.55 ng/ml (0,15-2,25) , DHEAS 133,10 µg/dl (150-210), progesterona 3,14 ng/ml (0,05-0,8), 11 deoxicortisol 20,4 ng/ml (0-30), testosterona total 0.34 ng/ml (0,05-4,15), dihidrotestosterona 0,17 ng/ml (0,12- 0,85), LH 1.7 mUI/ml (0-7,07) , FSH 2,20 mUI/ml (0,20-1,7), cortisol 13,97 µg/dl (2,8-23), ACTH 84.5 pg/ml (0-161), aldosterona 133 ng/dl (2-70), actividad renina plasmática 82,8 ng/ml/h (0,6-21,3), SHBG 109 nmol/l (60-250), hormona antimülleriana 474 pmol/l (251-679). No se pudo determinar 17OHpregnenolona y DHEA. El test corto de hCG mostró una escasa respuesta de testosterona y el test de ACTH una respuesta adecuada de cortisol con aumento de hormonas suprarrenales. Las pruebas de imagen y la laparoscopia reflejaron la existencia de testículos en conducto inguinal, ausencia de estructuras müllerianas y presencia completa de estructuras wolffianas. La genitografía mostró un seno urogenital con vagina de fondo ciego de 1,5 cm. Se realizó biopsia gonadal con presencia de tejido testicular morfológicamente normal. El estudio molecular del gen HSD3B2 reveló heterocigosis compuesta para las mutaciones c.244G>A (p.Ala82Thr) (A82T) (previamente descrita) y la nueva mutación c.1016A>G (p.Tyr339Cys). Conclusión: En el déficit de 3βHSD2, a pesar de existir un bloqueo en la conversión de delta-5 esteroides a delta-4esteroides, podemos encontrar cifras elevadas de 17 OHP debido a la conversión periférica realizada por la isoenzima tipo 1 (HSD3B1). Nuestro caso demuestra el efecto beneficioso del screening neonatal de 21 OH para la detección de patologías suprarrenales menos frecuentes. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites