| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):252-256 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.196 |
| Displasias óseas |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
 Figura - P2/d3-174 Figura - P2/d3-174 |
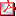 Figura - P2/d3-175 Figura - P2/d3-175 |
|
|
P2/d3-172 ACRODISÓSTOSIS Y PSEUDOHIPOPARATIROIDISMO (PHP-Ia): ENFERMEDADES DIFERENTES CON CLÍNICA SIMILAR M. Ruiz Goikoetxea(1), A. Sagastibelza Zabaleta(1), S. Berrade Zubiri(1), M.J. Chueca Guindulain(1), G. Pérez de Nanclares Leal(2), M. Oyarzabal Irigoyen(1) (1) Complejo Hospitalario de Navarra, Pamplona; (2) Hospital Universitario de Álava, Vitoria Introducción: El PHP-Ia es un síndrome producido por mutaciones en el gen GNAS, que conduce a una disminución de la subunidad alfa de la proteína G estimulante, que da lugar a resistencia multihormonal (principalmente PTH y TSH) y fenotipo de osteodistrofia de Albright (baja talla, facies redonda, falanges cortas, acortamiento 4-5 º metacarpianos). Estas características clínicas y bioquímicas son similares a otra enfermedad de diferente origen genético denominada acrodisóstosis. Pacientes: En los últimos 25 años, nuestra unidad ha estudiado 4 niños con alteraciones óseas y valores elevados de PTH y TSH; 3 casos se diagnosticaron de PHP-Ia y 1 caso de acrodisóstosis: Se trata de una niña de 3 años y medio de edad remitida a consulta para estudio de talla baja. Antecedentes personales: Ingreso en Neonatología por hipoglucemia y CIR (-2,3 DE) Cariotipo (46 XX). Desarrollo psicomotor normal. Antecedentes familiares: sin interés. Exploración física: peso 14,5 kg (p35) y talla 91,3 p4 (-1,75 SDS), braquidactilia y raíz nasal deprimida. Pruebas complementarias: Rx serie ósea: huesos de las manos cortos y toscos; resto normal. Analítica: PTH 117,2 pg/mL, calcio 10,2 mg/dL, calcio iónico 4,8 mg/dL, fósforo 5,6 mg/dL, TSH 14,13 µUI/ mL y FT4 1,2 ng/dL. Se inició tratamiento sustitutivo con L-tiroxina. El estudio genético, tras resultado negativo para GNAS, evidenció uma mutación de novo en heterocigosis exon 9 del gen PRKAR1A, diagnóstico de acrodisóstosis. Conclusiones: • Las similitudes clínicas y bioquímicas entre el PHP-Ia y la acrodisóstosis pueden conducir a un diagnóstico erróneo. El conocimiento de esta entidad permite la correcta identificación de estos niños, en los que no se encuentran mutaciones para GNAS. • La mutación en PRKAR1A supone una resistencia de una proteína kinasa A a la estimulación del AMPc, causando displasia ósea congénita y resistencia multihormonal.
P2/d3-173
DISPLASIA ÓSEA POR DELECCIÓN 13Q31.1Q 31.2. L. Rey Cordo, S. Pereiro Fernández, J.L. Chamorro Martín, A. Repáraz Andrade, C. Melcón Crespo, D. González Lestón Complejo Hospitalario Universitario de Vigo (CHUVI)/Servicio de Pediatría, Vigo/Pontevedra Introducción: Las displasias esqueléticas plantean un reto diagnóstico en la Consulta de Endocrinología Pediátrica. La nomenclatura internacional y clasificación de las osteocondrodisplasias está sujeta a debate debido a los avances en Biología Molecular de los últimos años. Presentamos un caso de omodisplasia por delección del gen GPC6 en heterocigosis. La Omodisplasia es una displasia esquelética infrecuente caracterizada por dismorfia facial y acortamiento de miembros. Caso clínico: Paciente de 12 meses de edad remitido a la Consulta de Endocrinología Pediátrica para estudio de talla baja. Antecedentes personales: ingresado al nacimiento por arritmia fetal. Antecedentes familiares: talla baja en rama materna, retraso constitucional del crecimiento en rama paterna. Exploración física: facies peculiar con abombamiento frontal, angioma plano frontal en línea media, hipertelorismo, raíz nasal aplanada, boca triangular y orejas de implantación baja; manos cortas y globulosas y criptorquidia bilateral. Presenta un desarrollo psicomotor adecuado a su edad. La talla se encuentra en -4.17 DE respecto a la población de referencia, el peso en -2.76 DE y el perímetro cefálico en -3.09 DE. Exploraciones complementarias: analítica con anticuerpos antitransglutaminasa y funciones tiroidea, gonadal y somatotropa dentro de la normalidad. Serie ósea: se objetiva acortamiento de huesos largos, especialmente a nivel proximal (rizomelia). Cariotipo: se objetiva delección del cromosoma 13 (q31.1, q32.2), realizándose CGH array que evidencia una deleción heterocigota en la citobanda 13q31.1q31.2, coordenadas genómicas chr13: 81,728,515-99,596,963 de aproximadamente 17,87 megabases. Conclusiones: La omodisplasia es una enfermedad extremadamente infrecuente que debe ser considerada en los casos de talla baja y facies peculiar. La realización de cariotipo en los niños con talla baja puede ser de gran ayuda en al diagnóstico.
P2/d3-174
DISPLASIA ESPONDILOEPIMETAFISARIA CON HIPERLAXITUD ARTICULAR TIPO 1 L. Gutiérrez Pascual, M.A. Molina Rodríguez, L. Salamanca Fresno, J. Guerrero Fernández, I. Pastor Abascal, I. Gónzalez Casado Servicio Endocrino Infantil; Hospital la Paz. Madrid La displasia espondiloepimetafisaria con hiperlaxitud articular tipo 1 (SEMDJL1) es una displasia esquelética poco frecuente, de herencia autosómica recesiva, caracterizada por talla baja e hiperlaxitud articular generalizada que conduce a una deformidad espinal intratable con cifoescoliosis severa progresiva que puede comprometer la función respiratoria. Muchos niños tienen un rostro ovalado con ojos prominentes, escleróticas azules, y surco nasolabial largo. También se han descrito anomalías palatales y defectos cardíacos congénitos. Al nacimiento suelen presentar luxación de caderas y deformidades en pies. Presentamos a una niña de 6 años y 6 meses remitida desde el Servicio de Traumatología para valoración de síndrome malformativo. Al nacimiento se objetiva una luxación congénita de caderas, astrágalo vertical del pie izquierdo y pie equino derecho. Va desarrollando una escoliosis tóraco-lumbar de inicio precoz, progresiva. A la exploración presenta peso en -2,38 DE y talla en -5,39 DE; la cara es ovalada con frente amplia, ojos prominentes con escleras azuladas, labio superior fino y paladar ojival. El tórax es ancho y asimétrico, con una escoliosis tóraco-lumbar severa de convexidad izquierda e hiperlordosis lumbar muy marcada. Llama la atención una hiperlaxitud generalizada, con dificultad para la bipedestación (ver imagen). En la serie ósea se objetiva la escoliosis ya descrita, con vértebras ovoideas, una displasia acetabular con cabezas femorales luxadas y displásicas, y alteraciones metafisarias en miembros inferiores. Aporta estudio cardiológico y renal realizado al nacimiento, sin alteraciones. El nivel de compresión es normal. El tratamiento ortopédico y quirúrgico resulta habitualmente ineficaz dada la hiperlaxitud ligamentosa. La mayoría de los pacientes fallece entre la primera y segunda década de la vida por problemas compresivos y cardiorrespiratorios. Se han estudiado varios genes candidatos en casos familiares, como el COL1A1 , COL1A2 , COL2A1 , la fibrilina o la elastina, y aunque sabemos que la herencia es autosómica recesiva, el defecto molecular de la SEMDJL1 no ha sido identificado a día de hoy.
P2/d3-175
DISPLASIA MESOMÉLICA DE LANGER: EXPRESIÓN CLÍNICO-RADIOLÓGICA DE UN CASO M.A. Molina Rodríguez, AC. Barreda Bonis, I. Pastor, V. Barca-Tierno, K. Heath, I. González-Casado Hospital Infantil La Paz/Servicio de Endocrinología Infantil, Madrid Introducción: La displasia mesomélica de Langer (DML) es un enanismo micromélico fruto de la nula expresión funcional del gen SHOX. Existen pocos casos descritos en la literatura, presentándose en la mayoría de los casos, en familias con fuerte carga de consanguinidad, y en nuestro medio, en etnia gitana. Caso clínico: Lactante de etnia gitana de 6 meses que ingresa por hipoglucemia cetósica. Fruto de 2º embarazo, no controlado hasta la semana 32, detección de miembros cortos en 7º mes. Cesárea por pérdida del bienestar fetal en semana 39: peso 3.235 gr (p55, 0,13DE), longitud: 44 cm (p<1, -3,27DE) y perímetro cefálico: 35cm (p69, 0,52DE). Antecedentes familiares: madre 22 años, talla 149 cm (-2,51DS), deformidad de Madelung (Discondrosteosis de Leri-Weil: DLW). Padre 26 años, talla 165 cm (-1,92DS). Hermana 8 años, talla 125 cm (-0,62DS), sana. Tío por rama paterna con DML (talla 125 cm, -8,2DS), sordomudo. Tío rama paterna sordomudo. Consanguinidad entre abuelos. Deformidad de Madelung presente en abuela por rama materna y su hermana. Exploración física: 6 meses: Peso: 6,4kg (p10, -1.33DE); Longitud: 56 cm (p<1, -3.97DE); PC: 43,5 cm (p64, 0,36DE); Braza: 48 cm. Ratio de envergadura/longitud: 0,85. Llamativo acortamiento mesomélico y rizomélico. Tanner I. Pruebas complementarias: Serie ósea del caso índice: maxilar inferior hipoplásico, iliacos e isquión pequeños, acetábulos sin configurar. Acortamiento de fémures, tibias y marcadamente del peroné. Cavidad glenoidea sin configurar. Acortamiento de húmero, cúbito corto y trapezoidal y radio. Serie ósea. de la madre: deformidad bilateral de Madelung, exóstosis en fémur distal y tibia proximal (hallazgo). Estudio del gen SHOX: c.508G>C (p.A170P) en homocigosis en el índice y en heterocigosis en los padres. Conclusión: El cambio en A170P en el gen SHOX que presenta esta familia es la mutación más frecuente en etnia gitana e implica la inhibición del transporte de SHOX intranuclearmente, anulando su función. El estudio genético de DML o DLW en etnia gitana ha de iniciarse mediante el análisis de dicha mutación. Se postula su origen en un antecesor común.
P2/d3-176
PRESENTACIÓN DE 3 CASOS DE ACONDROPLASIA DE RECIENTE DIAGNÓSTICO M. García Sánchez, G. Martínez Moya, M. De Toro Codes, V. Esteban Marfin, J. De la Cruz Moreno Complejo Hospitalario de Jaén, Jaén Introducción: La acondroplasia es la forma más frecuente de displasia esquelética, afectando a la osificación encondral. Se caracteriza por talla baja, acortamiento rizomélico de extremidades, macrocefalia, raíz nasal deprimida, nalgas prominentes por hiperlordosis lumbar, mano en tridente. La clínica se acentúa con la edad. Presenta un patrón de herencia autosómica dominante, aunque en 80-90% de los casos se produce mutación de novo. El gen responsable se localiza en 4p16.3, y codifica el receptor 3 del factor de crecimiento fibroblástico (FGFR3). Las mutaciones G1138A y G1138C suponen el 99%. Objetivos: Presentamos 3 recién nacidas con diagnóstico prenatal de acondroplasia (estudio molecular de líquido amniótico: mutación c.1138G>A en el gen FGFR3). Casos Clínicos: Caso 1: Segunda gestación de madre de 36 años sana. Ecografía prenatal semana 34: acortamiento de fémur (41 mm) y húmero (39 mm). Parto a las 37 semanas. Peso 2,790g (-0,05sds) Longitud 43 cm (-2,46sds) Perímetro cefálico (PC): 35 cm (+1,05sds) Raíz nasal amplia, orejas de implantación baja, acortamiento rizomélico de extremidades, hipotonía axial discreta. Caso 2: Segunda gestación de madre de 37 años sana. Ecografía prenatal semana 37+3: acortamiento de fémur (47,6 mm) y húmero (46,5 mm). Parto a las 41 semanas. Peso 3.100 g (+0,53 sds) Longitud 45 cm (-1,75 sds) PC: 35,5 cm (+1,24 sds) Nariz en silla de montar, fontanela anterior amplia, acortamiento rizomélico de extremidades. Leve hipotonía axial. Caso 3: Primera gestación de madre de 26 años sana. Ecografía prenatal semana 29+5: acortamiento de fémur (43 mm) y húmero (40 mm). Confirmándose diagnóstico en semana 36. Parto a las 39 semanas. Peso: 3.330g (+0.39 sds) Longitud: 49 cm (-0.26 sds) PC: 37 cm (+1.9 sds) Frente abombada, acortamiento rizomélico de extremidades. Leve hipotonía axial. Pruebas complementarias: Mapa óseo: Aspecto tosco de metáfisis proximal de fémures y húmeros, forma cuboidea de vértebras, aumento de espacios discales intervertebrales y bordes irregulares de ambos ilíacos. Ecografía transfontanelar: normal. Conclusiones: Aunque su frecuencia en la población es baja, en nuestro centro se han registrado 3 casos en los últimos 18 meses. A pesar de los avances en medicina fetal, su diagnóstico temprano sigue siendo un reto. El asesoramiento genético puede ayudar a tomar decisiones bien fundamentadas relativas a la planificación familiar.
P2/d3-177
ESTUDIO DESCRIPTIVO DE LOS CASOS DE OSTEOGÉNESIS IMPERFECTA DIAGNOSTICADOS EN NUESTRO CENTRO J.L. Chamorro Martín, L. Caride López, D. Cañizo Vázquez, A. Collazo Álvarez, S. Pereiro Fernández, C.L. Rey Cordo Complejo Hospitalario Universitario de Vigo/ Servicio de Pediatría, Vigo/ Pontevedra Introducción y Objetivos: La osteogénesis imperfecta abarca un grupo de enfermedades hereditarias que afectan a la producción de colágeno. Clínicamente se caracteriza por fragilidad ósea, talla baja, escleras azules, dentinogénesis imperfecta, hipermovilidad articular, sordera en la edad adulta y una amplia variabilidad clínica. El objetivo de nuestro estudio es realizar una revisión de las características clínicas y la evolución de los pacientes diagnosticados en nuestro Centro. Material y métodos: Estudio descriptivo retrospectivo de los pacientes diagnosticados de osteogénesis imperfecta en nuestro Centro desde el 1 de Enero de 2006 al 31 de diciembre de 2012. Discusión: Se han diagnosticado siete casos de osteogénesis imperfecta. La media de edad al diagnóstico fue de 4.5 años. Todos los pacientes cumplen criterios clínicos de esta enfermedad: la fragilidad ósea y las escleras azules están presentes en el 100%. Seis pacientes recibieron tratamiento con Pamidronato según protocolo de Plotkin de Montreal modificado. Tres no presentaron nuevas fracturas tras el inicio del tratamiento, dos presentaron algún tipo de fractura posterior. El sexto paciente presentó mala adherencia al tratamiento, produciéndose múltiples fracturas a lo largo de la evolución de la enfermedad. El séptimo caso todavía no ha iniciado tratamiento con Pamidronato. Todos los pacientes se encontraban en el momento del diagnóstico por debajo de la talla estimada respecto a la población de referencia para su edad y sexo. En todos los pacientes que recibieron tratamiento con Pamidronato se observó mejoría en la talla a lo largo de la evolución de la enfermedad. Esta mejoría también fue objetivable en las densitometrías pre y post-tratamiento. En cuanto al estudio genético, se encuentra pendiente de resultado en tres pacientes. Se objetivó mutación del gen COLA1 en dos pacientes. En dos no se objetivó mutación del gen, estando pendiente el estudio del COLA2. El estudio fue negativo en un caso. Conclusiones: La osteogénesis imperfecta es una enfermedad infrecuente. El tratamiento con Pamidronato parece disminuir el número de fracturas en nuestra serie, lo que supone una mejora de la calidad de vida y del pronóstico de movilidad. Un inicio del tratamiento precoz permitirá reducir las secuelas óseas de la enfermedad. |
| References |