
| Rev Esp Endocrinol Pediatr 2014;5 Suppl(1):96-112 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2014.Apr.231 |
| Crecimiento, metabolismo y nutrición, diabetes, tiroides, suprarrenales, gónadas, hipotálamo-hipófisis, genética, displasias óseas |
| Sent for review: 25 Apr. 2014 | Accepted: 25 Apr. 2014 | Published: 8 May. 2014 |
| SEEP |
| Correspondence: SEEP E-mail: seep@seep.es |
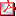 Tabla O1/d2/010 - Mutaciones IGF1R Tabla O1/d2/010 - Mutaciones IGF1R |
| Tabla O1/d2/016 - IHC |
 Tabla O1/d2/017 - DM1 Tabla O1/d2/017 - DM1 |
| Tabla O1/d2/018 - Tratamiento ISCI largo plazo |
| Tabla O1/d2/020 - Mutaciones digénicas MODY |
| Tabla O1/d2/021 |
| Tabla O1/d2/025 - HSC-NC |
| Tabla O1/d2/026 - HSCNC.TEST ACTH |
| Tabla O1/d2/027t |
| Tabla O1/d2/029 - Pubertad Precoz Central RM mujeres |
| Tabla O1/d2/030 - Turner-Calidad de vida |
| Tabla O1/d2/031 - SPW |
|
|
Crecimiento
O2/d2-009 IDENTIFICACIÓN DE MUTACIONES EN EL GEN NPR2 EN PACIENTES CON TALLA BAJA A. Hisado Oliva 1, S. Benito Sanz 1, A. Belinchón 1, E. Vallespín 1, A. del Pozo 1, A.C. Barreda Bonis 2, J Ramírez 3, C. Luzuriaga 4, I. González Casado 2, A. Campos Barros 1, K.E. Heath 1.
(1) Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Universidad Autónoma de Madrid (UAM), IdiPAZ y CIBERER, ISCIII. Madrid. (2)Servicio de Endocrinología Pediátrica, Hospital Universitario La Paz, UAM. Madrid. (3) Servicio de Endocrinología Pediátrica, Hospital Universitario Príncipe de Asturias. Alcalá de Henares, (4) Servicio de Endocrinología Pediátrica, Hospital Universitario Marqués de Valdecilla, Santander.
Introducción: La discondrosteosis de Léri-Weill (DLW) se caracteriza por talla baja desproporcionada debido a un acortamiento mesomélico de las extremidades y por la deformidad de Madelung. Aproximadamente el 70% de los casos se pueden explicar por alteraciones del gen SHOX o sus regiones reguladoras, mientras que se desconoce el defecto molecular del 30% restante. Recientemente, se identificaron mutaciones en heterocigosis en el gen NPR2 (Natriuretic Peptide Receptor-2) en 3 pacientes con talla baja idiopática (6%), uno de ellos presentaba talla baja desproporcionada. Cohorte y Métodos: Nuestro objetivo fue buscar si mutaciones en el gen NPR2 están presentes en nuestra cohorte de pacientes con DLW o sospecha de tenerla sin defecto molecular conocido (n=87). La búsqueda de mutaciones se hizo mediante secuenciación masiva (Next-generation sequencing) usando un panel de genes implicados en displasias esqueléticas (HaloPlex, Agilent) y la plataforma MiSeq (Illumina) o mediante High resolution melting y secuenciación. El análisis de la patogenicidad de los cambios se realizo usando el software Alamut V2.3-6.
Resultados: Hemos identificado cuatro nuevas mutaciones en heterocigosis en NPR2: exón 10: c.1641_1643del (p.Val548del); exón 19: c.2759G>A (p.Gly920Asp), exón 20: c.2972A>G (p.Glu991Gly) y una en exón 21: c.3058C>T (p.Arg1021Trp). Estos cambios no han sido observados en ningún control de 1.000 genomes o Exome Variant Server. El análisis in silico de ellas indica que los cuatro cambios afectan a aminoácidos altamente conservados y nos sugiere que las tres mutaciones sin sentido son probablemente patogénicas, por otro lado la patogenicidad de la deleción es desconocida. Además hemos confirmado la cosegregación de la mutación p.Glu991Gly en los tres miembros familiares afectados.
Conclusiones: 1) Mutaciones en heterocigosis del gen NPR2 han sido identificadas en 4,6% de los pacientes de nuestra cohorte de DLW o sospecha de tenerla; 2) Dos de los cuatro probandos y familiares de los otros dos presentan talla baja desproporcionada pero con ausencia de Madelung; 3) En un futuro llevaremos a cabo estudios de cosegregación y funcionales para demostrar la patogenicidad de los cambios encontrados; 4) Aconsejamos la búsqueda de mutaciones en el gen NPR2 en pacientes con talla baja desproporcionada o DLW sin defectos en SHOX.
O2/d2-010 MUTACIONES EN HETEROZIGOSIS DE IGF1R REPRESENTAN UNA CAUSA FRECUENTE DE HIPOCRECIMIENTO ARMÓNICO ASOCIADO A RESISTENCIA PARCIAL A IGF1 A. Campos Barros1, A. Gómez Núñez1, I. González Casado2, L. Salamanca Fresno2, S. García Miñaur1, P. Ros Pérez3, MV. Borrás Pérez4, L. Audí Parera5, J. Rosell Andreo6, D. de Sotto Martín7, B. García Cuartero8, A. González Vergaz8, J. Cruz Rojo9, L. Garzón9, ME Gallego Gómez9.
(1)Hospital Universitario La Paz, INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, y CIBERER (U753), ISCIII, Madrid. (2) Servicio de Endocrinología Pediátrica, Hospital Universitario La Paz, Madrid. (3) Servicio de Pediatría, Hospital Universitario Puerta de Hierro-Mahadahonda, Madrid (4) Endocrinología Pediátrica, Hosp. General de Granollers, Barcelona. (5) Servicio de Pediatría, Hosp. Vall de Hebrón, VHIR, CIBERER (U712), ISCIII, Barcelona. (6) Sección de Genética, Hosp. Universitari, Son Espases, Palma de Mallorca. (7) Endocrinología Pediátrica, Hosp. Universitari Son Espases, Palma de Mallorca. (8) Servicio de Endocrinología Pediátrica, Hosp. Universitario Severo Ochoa, Madrid. (1) Servicio de Endocrinología Pediátrica, Hosp. Universitario 12 de Octubre, Madrid.
El síndrome de resistencia a IGF1 (SR-IGF1) se caracteriza por retraso del crecimiento intrauterino y postnatal con niveles disminuidos, normales o supranormales de IGF-I e IGFBP3, microcefalia acompañada de rasgos dismórficos y déficit intelectual variable. SR-IGF1 está causado por anomalías genómicas o mutaciones que afectan al gen IGF1R (15q26.3), codificante del receptor de IGF1.
Objetivo: Investigar la incidencia de defectos moleculares en el gen IGF1R, en una cohorte de pacientes con hipocrecimiento armónico sin defecto molecular conocido.
Sujetos de estudio: 46 pacientes nacidos con bajo peso y talla para edad gestacional (SGA) y/o hipocrecimiento armónico postnatal sin defecto molecular conocido.
Estudios moleculares: Rastreo mutacional de las secuencias codificantes, transiciones intrón/exón y secuencias reguladoras de los genes IGF1 e IGF1R mediante HRM y secuenciación; análisis de deleciones/duplicaciones de la región 15q26.3 mediante MLPA y aCGH.
Resultados: Se identificaron un total de 8/46 (17,4%) pacientes con alteraciones en el gen IGF1R (ver Tabla 1). Dos presentaron deleciones de novo en heterozigosis de la región 15q26.2-26.3, que incluye a IGF1R. Los 6 restantes presentaron 5 mutaciones diferentes en heterozigosis (4 no descritas), 4 de las cuales afectan a aminoácidos filogenéticamente muy conservados, siendo la quinta una duplicación que altera la pauta de lectura, extendiendo y alterando la composición del extremo carboxiterminal de la proteína. Todas ellas fueron clasificadas como probablemente patogénicas por herramientas bioinformáticas para el análisis de variantes. Las principales características clínicas de los pacientes se resumen en la Tabla 1.
Conclusiones: Mutaciones en heterozigosis de IGF1R representan un defecto molecular relativamente frecuente (17,4%) en los pacientes nacidos SGA y/o con hipocrecimiento armónico postnatal estudiados. La manifestación fenotípica de las mutaciones es muy variable siendo predominantes el crecimiento intrauterino restringido, la microcefalia, el retraso del desarrollo psicomotor y los niveles elevados de IGF1, si bien estos no son uniformes en todos los pacientes con mutaciones identificados hasta la fecha. Estos resultados indican que mutaciones en heterozigosis de IGF1R pueden aparecer asociadas a un síndrome de hipocrecimiento armónico por resistencia parcial a IGF-I de características fenotípicas muy variables y prevalencia probablemente superior a la inicialmente prevista.
O2/d2-011 SUBUNIDAD ÁCIDO LÁBIL: VALORES DE REFERENCIA EN POBLACIÓN PEDIÁTRICA PREPUBERAL Y SU COMPARACIÓN CON NIÑOS CON TALLA BAJA DE DISTINTA ETIOLOGÍA P. González Ildefonso1, P. Prieto Matos1, P. Prieto Matos2, L. Regueras Santos3, S. Rupérez4, M. Martín Alonso1, A. Blanco Barrio5, M. Hernández Cerceño6, I. Mulero7,SI. de la Torre Santos8, E. Garcia Serrano1, MP. Sendín Hernández6, Grupo de trabajo de endocrinología pediátrica de castilla y león.
(1)Hospital Universitario Salamanca. (2) Hospital Clínico Valladolid. (3) Hospital de León. (4) Nuestra Señora de Sonsoles. Ávila. (5) Hospital Universitario Burgos. (6) Servicio de análisis clínicos. Hospital Universitario Salamanca. (7) Hospital Río Hortega. Valladolid.
Introducción: La subunidad ácido lábil (ALS) es una proteína que forma parte del complejo ternario de la hormona de crecimiento (GH) cuyo objetivo es aumentar la vida media de la IGF1. Su estudio es reciente por lo que no existen valores de referencia en niños ni sus variaciones en las distintas etiologías de talla baja.
Objetivo: Definir los valores normales de ALS estratificados por edad, sexo, edad ósea en los niños prepuberales de nuestra comunidad y describir la variabilidad de ALS en la talla baja de diferente etiología.
Pacientes y Métodos: Estudio multicéntrico prospectivo de casos (pacientes pediátricos prepuberales con talla baja) (n=95) y controles (paciente pediátricos prepuberales con talla normal) (n=33 recogiendo niveles de ALS, diagnóstico, antropometría, estadio puberal, edad ósea y diversos valores analíticos.
Resultados: La media de ALS de los controles es de 1779,1 mU/ml (1127-1930). En el grupo control se encuentra correlación significativa de ALS con edad (r=0,423; p=0,017) y con insulina (r=0,400; p=0,043), sin encontrar correlación con glucosa (p=0,531) ni IGF1 (p=0,271). Si incluimos en el estudio los pacientes con talla baja y los controles persiste la correlación significativa con edad (p<0,001; r=0,487) e insulina (p=0,007; r=0,289) apareciendo correlación positiva con IGF1 (p<0,001; r=0,600) y con glucosa (p=0,002; r=0,284). En el grupo de niños con talla baja el 14,8% son pequeños para su edad gestacional (PEG), el 16,8% tienen déficit de GH (DGH), el 18,9% presentan un retraso constitucional del crecimiento y desarrollo (RCCD), el 24,2% tienen talla baja familiar (TBF) y el 25,3% talla baja idiopática (TBI).
Se comparan los valores de ALS de los pacientes controles con los distintos grupos de talla baja objetivando niveles más elevados respecto a los pacientes con déficit de GH (p=0,036) sin encontrar diferencias significativas respecto a pacientes pequeños para su edad gestacional (p=0,411), retraso constitucional del crecimiento y del desarrollo (p=0,938), talla baja familiar (p=0,991) y otras tallas bajas idiopáticas (p=0,084).
Conclusiones: Los niveles de ALS están disminuidos en niños prepuberales con DGH. No se encuentran alteraciones en los niveles de ALS en pacientes prepuberales con talla baja secundaria a PEG, RCCD, TBF u otras TBI.
O2/d2-012 Evaluación de la edad de retirada del tratamiento con análogos de GnRH, la edad de la menarquia y la talla adulta en niñas con Pubertad Precoz Central (PPC) en el registro PUBERE de la Sociedad Española de Endocrinología Pediátrica (SEEP) R. Corripio Collado1, L. Soriano Guillén2, R. Cañete Estrada3, L. Castro Feijoó4, A. Escribano Muñoz5, R. Espino Aguilar6, J. Herreo Espinet7, JI. Labarta Aizpún8, J. Argente Oliver9. (1) Hospital de Sabadell. Corporació Sanitària Parc Taulí/ Unidad Endocrinología Pediátrica. (2) Hospital Fundación Jiménez Díaz, Madrid. (3) Hospital Universitario Reina Sofía, Córdoba. (4) Hospital Clínico Universitario de Santiago de Compostela. A Coruña. (5) Hospital Universitario Virgen de la Arrixaca, Murcia. (6) H. Universitario Virgen de Valme, Sevilla. (7) Hospital St. Jaume de Calella, Barcelona, (8) Hospital Miguel Servet, Zaragoza, (9) Hospital Universitario Niño Jesús, Madrid.
En el año 2007 se creó el Registro Español PUBERE con 53 hospitales. Entre sus objetivos: a) conocer la edad cronológica (EC) y ósea (EO) de la retirada del tratamiento con análogos de GnRH; b) analizar la edad de la menarquia y tiempo transcurrido tras retirar el tratamiento; c) conocer los datos de talla adulta.
Criterios de inclusión: Pacientes con PPC nacidos a partir de 1992, diagnosticados antes de los 8 años de edad en niñas y de los 9 en niños, con EO > EC y pico de LH tras LHRH > 7 U/L.
Objetivos: -Talla adulta y predicción de talla adulta (PTA) al diagnóstico mediante (Bayley-Pineau) average girls (cm y SDS) respecto a población de referencia. -Interrupción del tratamiento (EO y EC), edad de la menarquia y talla.
Resultados: Se incluyen 422 pacientes (91,5 % niñas, 19,9 % adoptados, 87.4 % idiopáticos); con datos de fin de tratamiento (n=186 niñas), edad de menarquia (n=106) y talla adulta (n=30 niñas). La edad al inicio del tratamiento (♀) fue 7,26 ± 1,31 años, la EO 9,1 ± 1,51 años. La talla adulta fue de 157,5 ±11,03 cm (-0,66 ± 1,98 SDS) respecto a la población de referencia. La PTA al diagnóstico fue de 161,5 ± 10,9 cm. La diferencia entre la talla estimada al diagnóstico y la final fue de -3,76 ± 8.61 cm (IC95%: -6,98 a -0,55 cm) (p=0,023). La EC media de interrupción del tratamiento fue 10,15 ± 1,01 años, coincidiendo con una edad ósea de 11,6 años ± 0,67 años. El incremento de talla tras la interrupción del tratamiento fue de 13,1 cm ± 4,3 cm. La edad de la menarquia fue 11,04 ± 1,14 años; es decir, a los 13,52 ± 7,14 meses de interrumpir el tratamiento. Conclusiones preliminares: -De forma global, la talla adulta de las pacientes afectas de PPC fue significativamente inferior a la prevista al diagnóstico. -La interrupción del tratamiento se realiza antes de la edad de inicio puberal medio en la población general -La menarquia se produce unos 13 meses tras interrumpir el tratamiento, coincidiendo con datos referidos de otros estudios.
Metabolismo y Nutrición
O2/d2-013 EXPRESIÓN DE STK11 EN TEJIDO ADIPOSO TRAS RESTRICCIÓN DE PESO FETAL: RELACIÓN CON EL CRECIMIENTO RECUPERADOR Y LA MASA GRASA VISCERAL. G. Carreras Badosa 1, X. Remesar Betlloch 2, A. Prats Puig 1, L. Ibáñez Toda 3, J. Bassols Casadevall 1,
(1)IDIBGI, Hospital Dr. Josep Trueta de Girona.
Introducción: El gen STK11 regula el metabolismo de la glucosa y de los lípidos. Los ratones adultos con baja expresión de STK11 en tejido adiposo desarrollan un exceso de grasa y un perfil metabólico desfavorable. Desconocemos si una baja expresión de STK11 en tejido adiposo está relacionada con el crecimiento recuperador y el acúmulo de grasa tras restricción de peso fetal.
Objetivos: Estudiar la expresión de STK11 en tejido adiposo y su relación con el aumento de peso y de masa grasa visceral en ratas con restricción de peso fetal que presentan o no crecimiento recuperador postnatal.
Materiales y Métodos: Se estudiaron crías de ratas Wistar jóvenes en un modelo de restricción de crecimiento intrauterino inducido por restricción calórica durante el embarazo. Las ratas control parieron crías normales (C) y las restringidas, crías con bajo peso (R). Las crías de bajo peso manifestaron un crecimiento recuperador postnatal con dieta estándar (RC) o se mantuvieron delgadas con restricción calórica adicional después del nacimiento (RR). Se cuantificó a los 42 días de vida (10 crías de cada grupo) el aumento de peso postnatal, el depósito de masa grasa visceral (mesentérica, perigonadal y retroperitoneal) y la expresión de STK11 en tejido adiposo retroperitoneal.
Resultados: Las crías RC presentaron mayor incremento de peso postnatal a P42 en comparación con las crías RR y las ratas control (p<0.0001). Las crías RC presentaron mayor porcentaje de masa grasa visceral (p<0.0001) y de tejido adiposo retroperitoneal (p<0.0001) y menor expresión de STK11 en tejido adiposo retroperitoneal que las ratas RR (p<0.005). En ratas RC, la expresión de STK11 se asoció negativamente con el incremento de peso postnatal (r= −0,652, p<0.012), con la masa grasa visceral
Conclusión: La expresión de STK11 en tejido adiposo retroperitoneal se asocia al crecimiento recuperador postnatal y al depósito de masa grasa visceral en ratas con restricción de peso fetal. La menor expresión de STK11 puede ser uno de los mecanismos responsables del crecimiento recuperador postnatal y de la acumulación de tejido adiposo secundarios a restricción de peso fetal.
O2/d2-014 EL AUMENTO DE PESO Y DE TEJIDO ADIPOSO PRODUCIDO POR LA GHRELINA SE ASOCIA CON UN INCREMENTO EN LA SÍNTESIS DE CITOQUINAS EN EL HIPOTÁLAMO, SIN GENERAR GLIOSIS E. de la Fuente Martín, C. García Cáceres, M. Granado, F. Díaz, J. Argente, J.A. Chowen.
Hospital Infantil Universitario Niño Jesús, Instituto de Investigación la Princesa, Universidad Autónoma de Madrid, CIBERobn, Madrid
Introducción: La ghrelina es un péptido endógeno que modula la homeostasis energética, estimulando el apetito e incrementando el tejido adiposo. Aunque la acumulación de grasa se asocia con la producción de mediadores inflamatorios, ambas isoformas de la ghrelina, acilada (GA) y desacilada (GD), ejercen efectos antiinflamatorios. Dado que los procesos inflamatorios están implicados en el desarrollo de complicaciones secundarias asociadas a la obesidad, como resistencia a insulina y leptina, la ghrelina podría proteger de las consecuencias negativas del incremento de ingesta.
Objetivo: Determinar si la ganancia de peso y la acumulación de tejido adiposo producidos por el tratamiento icv con GA o su análogo sintético, GHRP6, protegen frente a la inflamación y gliosis hipotalámica.
Material y Métodos: Se utilizaron ratas Wistar macho adultas, las cuales fueron implantadas icv con minibombas que infundían 5 ug/día de GA, GD, GHRP6 o salino (C), alimentadas ad libitum, y GA pairfed durante 14 días.
Resultados: La GA y el GHRP6 incrementaron el peso corporal (p<0,0001) debido a un aumento en la ingesta (p<0,0005), lo que correlacionó con un aumento en los niveles séricos de leptina (p<0,0001). Ambos tratamientos aumentaron el tejido adiposo visceral (p<0,0001), independientemente de la ingesta y sin generar un perfil pro-inflamatorio sistémico.
En el hipotálamo GA, y GD aumentaron la producción de citoquinas, pero no alteraron la expresión de proteínas de la vía de señalización de la leptina, como pSTAT3 o SOCS3, ni incrementaron la expresión de marcadores de gliosis, como GFAP, vimentina, CD68 o F4/80. Los tratamientos aumentaron la expresión de IGF-1, factor que estimula la neuroprotección.
Conclusión: El incremento de peso y de tejido adiposo producido por la ghrelina depende de su estado de acilación y se asocia con un incremento en la producción de citoquinas en el hipotálamo, sin generar gliosis ni activación de las vías de señalización implicadas en el desarrollo de resistencia a insulina y leptina. La ghrelina podría proteger frente al incremento de la ingesta mediante mecanismos que retrasen la gliosis hipotalámica, considerado un factor clave en la aparición de patologías metabólicas asociadas a la obesidad, uno de los cuales podría ser vía IGF-1.
O2/d2-015 EL ÁCIDO PALMÍTICO INDUCE UNA REDUCCIÓN DEL NÚMERO DE NEURONAS HIPOTALÁMICAS IN VITRO QUE ES REVERTIDA POR EL TRATAMIENTO CON IGF-1 A. Freire Fernández-Regatillo, F. Díaz, P. Argente, L.M. Frago, J. Argente, J.A. Chowen.
Hospital Infantil Universitario Niño Jesús, Instituto de Investigación la Princesa, Universidad Autónoma de Madrid, CIBERobn. Madrid.
Introducción: Se ha descrito un aumento de muerte celular en el hipotálamo de ratones obesos por la ingesta de una dieta rica en grasa. Además, los ácidos grasos saturados, como el ácido palmítico (PA), pueden ser tóxicos para las células. Este estudio trata de averiguar qué efectos podrían tener estos compuestos en el hipotálamo y si el IGF-1, un factor regulado por la nutrición, podría ejercer como neuroprotector ante esos daños.
Materiales y Métodos: Se ha empleado la línea neuronal hipotalámica de rata RCA-6, tratada durante 24 horas con diferentes concentraciones de PA, ácido oleico (OA) y una mezcla 1:1 de ambos. Para explorar un posible efecto neuroprotector del IGF-1, se ha hecho un pretratamiento con IGF-1 a diferentes concentraciones. La supervivencia celular se estimó con el método del cristal violeta. Mediante Western blot se han analizado los niveles de proteínas relacionadas con la apoptosis o la proliferación celular, como Bax, Caspasa-9 y PCNA.
Resultados: El tratamiento con PA a 0.1 mM provocó una reducción significativa del número de células. Los tratamientos con OA y la mezcla de OA + PA no afectó al número de células. El PA (0.5 mM) indujo una disminución de las proteínas Bax, y PCNA y un aumento de la Caspasa-9. Se observó una relación directa entre la concentración de PA y la disminución del número de células, siendo significativa a 0,5 mM. El IGF-1 (10 o 50 nM) contrarrestó el efecto del PA sobre la pérdida de neuronas.
Conclusión: Una dieta rica en grasas saturadas puede tener efectos perjudiciales incluso en el cerebro y pueden existir mecanismos neuroprotectores endógenos que contrarresten estos efectos. El papel neuroprotector del IGF-1 ya ha sido demostrado pero es interesante y novedoso conocer su efecto protector en dietas ricas en grasa. Finalmente, hay indicios de que el ácido oleico podría revertir los efectos deletéreos del ácido palmítico.
O2/d2-016 LA ALTERACIÓN EN LA SECRECIÓN DE ADIPONECTINA INFLUYE EN EL DESARROLLO DE INTOLERANCIA A LOS HIDRATOS DE CARBONO EN NIÑOS Y ADOLESCENTES OBESOS A. Bartucci 1, V. Barrios 1, J. Argente 2, G.A. Martos-Moreno 2.
(1)Hospital Infantil Universitario Niño Jesús. IIS La Princesa/Servicio de Endocrinología. ISCIII/CIBERobn. Madrid. (2) Hospital Infantil Universitario Niño Jesús. IIS La Princesa/Servicio de Endocrinología. UAM/Departamento de Pediatría. ISCIII/CIBERobn. Madrid.
La intolerancia a los hidratos de carbono (IHC, glucosa en test de tolerancia oral a glucosa [TTOG ]≥140mg/dl tras 120 minutos) en la obesidad infanto-juvenil está altamente influenciada por grado de exceso de peso, aunque también depende de factores predisponentes individuales.
Estudiar la influencia de la secreción de adiponectina total (T-ADP) y de alto peso molecular (HMW-ADP) en el desarrollo de IHC en niños y adolescentes obesos.
Se analizó comparativamente una cohorte de 87 pacientes obesos (IMC >+2SDS) con IHC (OB-IHC) y un grupo pareado (por edad, sexo, etnia e IMC-SDS) de pacientes obesos sin alteración del metabolismo HC (OB-sanos) (Tabla 1). Se registraron antecedentes familiares de diabetes, antropometría, presencia de acantosis nigricans, esteatosis hepática y niveles de HbA1c, Colesterol total, LDL, HDL, VLDL, triglicéridos, ácido úrico, glucemia e insulinemia basales y tras TTOG (1,75g/kg, máximo 75g), así como los índices derivados de ésta [HOMA, WBISI, QUICK, índice insulinogénico (I-I), área bajo de la curva (AUC) para glucemia e insulina] y se estudió su relación con los niveles circulantes de leptina (RIA), receptor soluble de leptina (R-LEP, ELISA), T-ADP (RIA) y HMW-ADP (ELISA).
El grupo OB-IHC, que presentaba mayores niveles de insulina y glucosa, basales y tras TTOG, I-I, HbA1c e índices de resistencia a insulina; mostraba también niveles superiores de leptina (p<0,001) e inferiores de T-ADP (p<0,05), HMW-ADP (p<0,01) y R-LEP (p<0,05); resultando superiores sus cocientes leptina/R-LEP y leptina/T-ADP (ambos p<0,001) (TABLA). Al realizar correlaciones parciales, controladas para IMC-SDS, se observó que los niveles de ADP-T correlacionaban negativamente con la glucemia a los 120´ en el TTOG (r = -0,253, p<0,01), HOMA (r = -0,253, p<0,01), AUC-insulina (r = -0,325, p<0,01) e I-I (r = -0,355, P<0,001). Análogamente, HMW-ADP se correlaciona negativamente con glucemia 120´(r = -0,305, p< 0,05), HOMA (r = -0,192, p<0,05), AUC-insulina (r = -0,245, p<0,05), I-I (r = -0,245, p<0,05) y positivamente con los índices de sensibilidad a insulina WBISI (r = +0,270, p<0,05) y QUICK (r = +0,237, p<0,05). La afectación de la secreción de adiponectina en niños y adolescentes obesos influye en el desarrollo de IHC, independientemente del grado de exceso de IMC.
Diabetes
O3/d3-017 IMPACTO DE LOS CONOCIMIENTOS DIABETOLÓGICOS DE LOS PADRES SOBRE EL CONTROL METABÓLICO DE LOS PACIENTES PEDIÁTRICOS CON DIABETES MELLITUS TIPO 1 DE. Alonso Martín, R. Yelmo Valverde, M.A. Álvarez Gómez, M. Martín-Frías, R. Barrio Castellanos, B. Roldán Martín.
Unidad de Endocrinología Pediátrica, Hospital Universitario Ramón y Cajal, Madrid.
Introducción: La educación diabetológica es el principal instrumento terapéutico para conseguir la implicación del entorno familiar de los pacientes pediátricos con diabetes mellitus tipo 1 (DM1).
Objetivo: Determinar si el nivel de conocimientos diabetológicos de los padres afecta al control glucémico valorado por la HbA1c.
Pacientes y Métodos: El cuidador principal y el adolescente en su caso rellenaron un cuestionario de conocimientos sobre diabetes de 14 ítems (Mitchell K, Diab Technol Ther 2013) adaptado a la modalidad de tratamiento: múltiples dosis de insulina (MDI) o sistema de infusión subcutánea de insulina (ISCI). Las 10 primeras preguntas valoran los conocimientos teóricos sobre la enfermedad, las otras 4 la sensación percibida sobre su grado de participación en los cuidados. Todas las familias/pacientes habían recibido educación diabetológica reglada al diagnóstico, al mes y en cada revisión clínica. Se recogen datos de la historia clínica, el nivel educativo de los padres y se utiliza la HbA1c media del último año.
Resultados: Se analizan 105 encuestas, 67 en tratados con MDI y 38 con ISCI. Edad media de los pacientes: 11,4±3,6 y 11,5±4,1años, respectivamente; HbA1c media de 6,6±0,5 y 6,5±0,5%. Las encuestas fueron contestadas por madres en un 68,7% en el primer grupo y por un 57,9% en el segundo. La puntuación media obtenida en conocimientos fue de 8,3 sobre 10 (grupo MDI) y de 7,5 (grupo ISCI). Un 62,7% de los encuestados en tratamiento con MDI y un 89,5% con ISCI consideran continuar aumentando sus conocimientos sobre DM. No se encontraron diferencias significativas al comparar el nivel de estudios de los cuidadores y la edad de los pacientes con el resultado de la puntuación objetiva de conocimientos ni con el nivel de HbA1c en los grupos analizados (Tabla). Tampoco la correlación entre la HbA1c y dicha puntuación fue significativa.
Conclusiones: 1. El alto nivel de conocimientos de los cuidadores se traduce en el buen control metabólico de nuestra población, independientemente del tipo de tratamiento. 2. La educación diabetológica continuada es fundamental para conseguir la motivación y la implicación de las familias en la consecución de los objetivos de tratamiento y el control glucémico.
O3/d3-018 TRATAMIENTO CON INFUSIÓN SUBCUTÁNEA CONTINUA DE INSULINA EN POBLACIÓN PEDIÁTRICA: EFICACIA Y SEGURIDAD A LARGO PLAZO M. Martín Frías, E. Colino Alcol, P. Enes Romero, M.A. Álvarez Gómez, R. Yelmo Valverde, R. Barrio Castellanos.
Hospital Ramón y Cajal, Madrid
Introducción: Existe poca información sobre la eficacia del tratamiento con ISCI durante periodos prolongados en la edad pediátrica.
Objetivos: - Valorar el control glucémico conseguido a largo plazo con el tratamiento con ISCI. - Analizar la seguridad de dicho tratamiento. - Valorar si la experiencia del equipo diabetológico afecta a los resultados.
Material y Métodos: Estudio retrospectivo en 90 pacientes (52 varones), menores de 18 años al inicio de la ISCI (edad media 10,1±4,5 años), que comenzaron durante la última década y que llevan al menos 1 año de tratamiento. Se excluyen aquellos pacientes con patología concomitante que pueda influir en el control de la DM1. Los pacientes y sus familias recibieron educación específica reglada. Se recogen las siguientes variables: edad al diagnóstico de la DM y en el momento del inicio de ISCI, estadio puberal, HbA1c (HPLC-Menarini 5,3 ± 0,41), necesidades de insulina (u/kg/día), porcentaje de insulina basal, ratios de insulina/hidrato de carbono (I/HC) en comidas, episodios de hipoglucemias graves (HG) (100 pacientes /año) y de cetoacidosis (CAD) y tasa de abandono del tratamiento. Se subdivide en 2 grupos según el año de inicio de la ISCI (grupo 1: 2003-2007, grupo 2: 2008-2012). El análisis estadístico se realiza mediante SPSS. Se comparan los datos mediante tests no paramétricos, siendo significativos si p<0,05.
Resultados: Al inicio de ISCI el 54% eran prepuberales. La HbA1c mejora al año y se mantiene posteriormente. Los episodios de HG graves y CAD disminuyen. La incidencia de HG durante el seguimiento de ISCI fue de 5,7 episodios/100 paciente-año. Sólo hubo 2 episodios de CAD. En el grupo 2 se objetivaron HbA1c menores y menos HG. La tasa global de abandono fue del 9% (Grupo 1: 13,6%, grupo 2: 4,3%). Resto de resultados en Tabla 1.
Conclusiones: - El tratamiento con ISCI es una alternativa eficaz y segura, a largo plazo, en el tratamiento de la DM1 en la edad pediátrica. Permite, aún en pacientes con buen control, mejorarlo y mantener en el tiempo dicha mejoría. - La experiencia del equipo diabetológico influye tanto en la selección de los pacientes como en los resultados.
O3/d3-019 ALTERACIONES DEL METABOLISMO HIDROCARBONADO Y FIBROSIS QUÍSTICA EN NIÑOS MENORES DE 10 AÑOS AB. Rodríguez Cejudo, I. Delgado Pecellín, A. Gómez Gila.
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción: El diagnóstico precoz de las alteraciones en el metabolismo de los hidratos de carbono (AMHC) en los pacientes con fibrosis quística (FQ) es fundamental, ya que la diabetes relacionada con la FQ se asocia con un empeoramiento del estado nutricional y/o función pulmonar. En la actualidad, se recomienda iniciar el cribado de AMHC a partir de los 10 años.
Objetivo: Evaluar la frecuencia de AMHC en pacientes con FQ menores de 10 años, así como su relación con otras complicaciones (respiratorias, insuficiencia exocrina, desnutrición) o su relación con mutaciones del gen CFTR y los índices insulinogénicos y HOMA.
Pacientes y Métodos: Estudio retrospectivo de 61 curvas de glucosa e insulina tras SOG, pertenecientes a 30 pacientes de entre 5-9 años con fibrosis quística. Variables: edad, sexo, índice de valoración nutricional , péptido C basal, HbA1C, IGF-1, valores de glucosa e insulina basales y a los 30, 60, 90 y 120 minutos, función pulmonar (CVF, VEF1 y CVF/VEF1), necesidad de enzimas pancreáticas, mutaciones CFTR, índice insulinogénico e índice HOMA. Estudio estadístico con programa SPSS.
Resultados: Edad media SOG: 7,5 años (5,05-9,88). 11 curvas glucosa patológicas (18,1% del total): 2 alteraciones de la tolerancia a glucosa ATG (3,3%), 9 alteraciones indeterminadas a la glucosa AIG (14,8%). Edad media AIG: 8,1 años, ATG: 8,76 años.
De todas las AMHC: 4 con desnutrición leve (36,36%), 1 sobrepeso y 6 con valoración nutricional normal (54,54%). 6 espirometrías del total de AMHC estaban alteradas (54,54%). Mutación CFTR más frecuente entre AMHC: F508del/F508del, seguida de F508del/ N1303K. Índice HOMA elevado en los dos grupos, valor medio normales 11,8 (9,84-13,86), valor medio AMHC 13,46 (10,28-16,65). No se han detectado diferencias entre índice insulinogénico de AMHC y curvas SOG normales.
Conclusiones: Se han encontrado AMHC antes de los 10 años, y es frecuente que se asocien a otras complicaciones, por lo que planteamos la necesidad de adelantar la edad del primer cribado. Más frecuencia de AMHC en mutaciones F508del/F508del. Ni la determinación de la HbA1c ni la necesidad de enzimas pancreáticas resultan de ayuda como método diagnóstico.
O3/d3-020 MUTACIONES DIGÉNICAS EN GCK Y HNF1A EN DIABETES MONOGÉNICA TIPO MODY A. Campos Barros 1, A. Gómez Núñez 1, JM Guardia Ortega 2, P Portillo Ortega 2, E. Caviola Mutazzi 3, M. Arráez Monllor 2.
(1) Hospital Universitario La Paz, INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, y CIBERER (U753), ISCIII, Madrid. (2) Servicio Endocrinología y Nutrición. Hospital Clínico Universitario Virgen de la Arrixaca. Murcia. (3) Endocrinología, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, Madrid.
Introducción: Mutaciones de pérdida de función en heterozigosis en los genes GCK y HNF1A constituyen la base molecular de la mayoría de los casos (20-65% en la población europea) de diabetes monogénica tipo MODY, siendo su prevalencia muy variable entre las diferentes poblaciones (0.6-2,2% de los casos de diabetes), conociéndose mas de 600 mutaciones de GCK y HNF1A.
Objetivo: Investigar la incidencia de mutaciones digénicas en GCK y HNF1A en una cohorte de 52 pacientes con diagnóstico molecular de diabetes monogénica tipo MODY, subtipo GCK.
Estudios moleculares: Rastreo de mutaciones en las secuencias codificantes, transiciones intrón/exón y secuencias reguladoras de los genes HNF1A y HNF4A mediante HRM y secuenciación. Genotipado de familiares de los pacientes portadores de mutaciones digénicas.
Resultados: Se identificaron dos casos con mutaciones digénicas, que presentaron, respectivamente, las mutaciones p.Asp271Asn en el exón 6 de GCK y p.Gly292Argfs25 en el exón 4 de HNF1A, en el primer caso, y p.Ile19Asn, en el exón 2 de GCK, y la mutación c.-64_-55delinsGC en el flanco 5 UTR (promotor) de HNF1A, en el segundo. Las principales características clínicas del paciente índice y familiares del primer caso se encuentran resumidas en la Tabla 1. Con respecto al caso 2, es una paciente diagnosticada a los 18 años, IMC 19,8 kg/m2; SOG: 109-116-172-128-106 mg/dl con antecedentes familiares en ambas líneas en tratamiento con Metformina.
Conclusión: A pesar de la predominancia de los subtipos GCK y HNF1A en la población europea, solamente se ha descrito un caso de DM tipo MODY con mutaciones digénicas, en GCK y HNF1A, en la literatura científica, por lo que los dos casos adicionales referidos nos aportan datos novedosos sobre su presentación, evolución clínica y respuesta a los tratamientos farmacológicos habituales. Es probable que la combinación de ambas mutaciones agrave el fenotipo clínico, por lo que es recomendable realizar un cercano seguimiento clínico de la evolución de los pacientes así como ampliar los estudios moleculares de aquellos con mutaciones en GCK que no respondan bien a las pautas dietéticas y recomendaciones de estilo de vida habituales.
O3/d3-021 MUTACIÓN EN ABCC8 Y HETEROGENEIDAD FENOTÍPICA L. Salamanca Fresno1, J. Guerrero Fernández1, C. Sevilla Arias1, A. Gómez Núñez2, I. González Casado1, A. Campos Barros3.
(1)Hospital Universitario La Paz/Servicio de Endocrinología Pediátrica, Madrid. (2) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz; Madrid. (3) INGEMM (Instituto de Genética Médica y Molecular), IdiPAZ, UAM, Hosp. Univ. La Paz y Centro de Investigación Biomédica en Enfermedades Raras (CIBERER, U753), Instituto Carlos III, Madrid.
Introducción: El hiperinsulinismo congénito (HI) es la causa más común de hipoglucemia persistente en la infancia. Las mutaciones en ABCC8 que codifica la subunidad SUR1 del canal de K suponen las formas más frecuentes y severas.
Material y Métodos: Se presentan 3 casos de HI no relacionados que comparten la mutación c.3574delG en el exón 29 de ABCC8 en heterozigosis, que introduce un codón stop prematuro: p.Asp1192MetfsX16. Estudios moleculares: Análisis mutacional de ABCC8 mediante HRM, secuenciación de variantes detectadas y análisis por MLPA de deleciones/duplicaciones.
Se analizan las características clínicas, fenotípicas, terapéuticas y evolutivas. Resultados: Las principales características se resumen en la Tabla 1. La presentación fue precoz en los casos 2 y 3, y más tardía en el 1 con crisis convulsiva. La pancreatectomía curativa fue necesaria en los casos 1 y 3, mientras que destaca la respuesta al diazóxido en el caso 2, que presentó datos de macrosomía al nacimiento (P75 en casos 1 y 3) y una discreta elevación de amonio (93 mmoles/L), con respuesta hipoglucémica en la sobrecarga oral de leucina. El desarrollo psicomotor es adecuado en todos, pero el 2 presentó crisis parciales con tratamiento antiepiléptico. Tan sólo la madre del caso 2 refiere datos de hipoglucemia.
Conclusiones: Se comprueba la elevada heterogeneidad del HI, incluso en pacientes que comparten la misma mutación. Aunque no se realizó estudio PET, la anatomía patológica mostró en el caso 1 una proliferación adenomatosa focal y en el 3 cambios difusos, que en ambos casos, sugieren la probable incidencia de un segundo evento: pérdida somática del alelo materno, en el primer caso y una segunda mutación en secuencias no codificantes o genes alternativos, en el segundo, todavía por identificar. Con respecto al caso 2, se han descrito previamente respuestas parciales a diazóxido en pacientes con mutaciones en ABCC8 en heterozigosis. La presencia de portadores sanos podría explicarse por una distinta penetrancia de la mutación ó también por la postulada presencia de un 2º evento por identificar, por ejemplo en GLUD1 en el caso 2 dados los antecedentes.
Tiroides
O3/d3-022 BOCIOGÉNESIS INTRAÚTERO EN EL HIPOTIROIDISMO POR DEFECTOS GENÉTICOS DE TIRO-GLOBULINA (TG) Y TIROPEROXIDASA (TPO). AM. Lucero1, A. Iglesias1, A.C. Barreda-Bonis1, P. Bahillo2, C. Luzuriaga3, G. Grau4, S. Rossignol5,T. Vu-Hong6, M. Polak7, I. González-Casado1, JC. Moreno1.
(1)Laboratorio Tiroides, INGEMM, Hospital Universitario La Paz. Madrid. (2) Endocrinología Infantil, Hospital Clínico Universitario de Valladolid. Valladolid.
El hipotiroidismo fetal con desarrollo de bocio intraútero es una enfermedad poco común que conlleva trastornos del desarrollo psicomotor y complicaciones obstétricas y neonatales graves.
Objetivo: Identificar la causa genética de bocios intraútero en nuestra serie de hipotiroidismo congénito (HC) e identificar porcentajes de detección ecográfica y actitud terapéutica en nuestro medio.
Pacientes y Métodos: Caracterización clínica, hormonal y genética de los pacientes. PCR y secuenciación Sanger de los genes de TG (exones con mutaciones frecuentes en caucásicos) y TPO. Secuenciación masiva (SM) de un panel de 18 genes tiroideos en 1 paciente.
Resultados: Identificamos 10 casos de bocio fetal de un total de 320 pacientes con HC. La TSH al nacimiento era muy elevadas (664 mU/L, 1 caso con 25.16 mU/L tras substitución oral de la madre con L-T4 durante la gestación), T4L bajas (0.4 ng/mL) y tiroglobulina muy baja (0.1-0.7 ng/dL, excepto un caso con 8.689 ng/dL).
En TG identificamos 4 mutaciones heterocigotas en 8 pacientes: dos codones-stop (p.R277X y p.R1511X) y dos missense (p.C1226Y, no descrita, y p.C1203Y). La SM detectó una deleción no descrita en el exón 16 de TG, críptica para la PCR.
En TPO identificamos 3 mutaciones en 2 pacientes: una homocigota (c.2422delT) en familia consaguínea que cambia el marco de lectura (p.C808fsX832) y, en otro paciente, una heterocigosis compuesta de dos mutaciones (p.G860R y p.R491C, no descrita). 7/10 casos tenían antecedentes familiares de hipotiroidismo o bocio, incluyendo 4 de hipotiroidismo gestacional. De los bocios detectados intraútero, solo 1/5 recibió inyección intraamniótica de T4 y 2/5 gestantes recibieron L-T4 oral. La posible asfixia neonatal requirió cesárea 2/2 casos. 1 paciente tiene grave retraso psicomotor a los 6 meses de edad.
Conclusiones: La prevalencia del bocio fetal es de 3,3% del HC, todos debidos a mutaciones en TG o TPO. Sólo 3 pacientes presentaron defectos bialélicos, lo que sugiere alteraciones crípticas que sólo detecta la SM. El tratamiento oral de la gestante con L-T4 no evita completamente el bocio en el HC. La inyección intraamniótica con T4 es inaceptablemente baja en nuestro medio.
O3/d3-023 Fenotipos clásicos y atípicos de resistencia a hormonas tiroideas N. Lacámara Ormaechea 1, B. Roldán 2, F. Morales 3, B. Barquiel 1, A. Escribano 4, I. Gonzalez-Casado 1, L. Vila 5, JC. Moreno 1.
(1)INGEMM - Hospital Universitario La Paz. (2) Hospital Ramón y Cajal. Madrid. (3) Hospital Nuestra Señora de Valme. Sevilla. (4) Hospital Universitario Virgen de La Arrixaca. Murcia. (5) Hospital Moises Broggi. Barcelona.
La resistencia a hormonas tiroideas (RHT) es la disminución de sensibilidad tisular a T3. Puede ser hipofisaria (sintomática) o generalizada (a/paucisintomática). En el 85% e identifican mutaciones en el gen THRB que codifica el receptor nuclear de T3, en el resto la etiología es desconocida. El ratón con deleción Dio2 expresa resistencia hipofisaria exclusivamente a T4.
Objetivo: Segregación clínica de pacientes con sospecha de RHT e investigación de su etiología molecular.
Métodos: PCR y secuenciación Sanger del gen THRB (exones 1-10) y de DIO2 (2/5 exones) en sospechas de resistencia hipofisaria a T4 exógena.
Resultados: En la cohorte de 45 pacientes (60% mujeres) con edad media de 23.5 años se distinguen 2 subgrupos: A. RHT clásica (n: 35 pacientes, 78%) con T4L y/o T3L elevadas y TSH no suprimida. La T4L está invariablemente elevada (2.69 ± 1.19 ng/dl; N: 0.9-1.7 ng/dl) y las TSH (6.43 ± 4.73 mU/L) en un 43%. Un 55% presentan clínica de hipertiroidismo. Se identifican mutaciones missense en heterocigosis en THRB en 16 pacientes (46%). Hay mutaciones frecuentes (R438H, P453T y R243W) y aisladas (R243Q, C309Y, R338W, M442V y K288R), todas localizadas en el dominio de unión al ligando. Tres casos asociaban adenomas hipofisarios y 3 autoinmunidad tiroidea. B. RHT hipofisaria atípica (n:10, 22%) sospechada por TSH elevada a pesar de altas dosis de L-tiroxina tras patología tiroidea primaria (agenesia tiroidea, tiroidectomía o ablación con yodo131 tras Graves o BMN -n: 6- o hipotiroidismo autoinmune -n:4-). Sus TSH son superiores a las del grupo A (13.11± 7.71 mU/L), con T4L normales (1.58± 0.4 ng/dl). El 70% presentan clínica hipertiroidea tras aumento de L-T4. Ninguno portaba mutaciones en TRHB o DIO2.
Conclusiones: Las RHT hipofisarias tras evento tiroideo primario (destrucción autoinmune, radioterápica o quirúrgica) no presentan perfil hormonal clásico de RHT pues la T4L está elevada sólo tras tratamiento sustitutivo con levotiroxina. Esta RHT atípica podría deberse a fallos hipofisarios de desyodasas. Excluyendo este grupo, el porcentaje de mutaciones de nuestra cohorte alcanza el 46%, similar al de cohortes internacionales. La RHT clásica puede asociar autoinmunidad tiroidea y formaciones adenomatosas hipofisarias que no corresponden funcionalmente con TSHomas.
O3/d3-024 TRIAC ENLENTECE EL DETERIORO NEUROLÓGICO EN EL SÍNDROME DE ALLAN-HERNDON-DUDLEY POR DEFECTO EN EL TRANSPORTADOR MCT8 DE HORMONAS TIROIDEAS A. Iglesias 1, A.L. Gómez-Gila 2, O. Alonso 2, J. A. Campos 3, M. Palomares 1, M. J. Obregón 4, B. Morte 4, J. Bernal 4, J. C. Moreno 1.
(1)Instituto de Genética Médica y Molecular (Ingemm) / Hospital Universitario La Paz. (2) Hospital Universitario Virgen del Rocío. Sevilla. (3) Hospital San Juan de Dios. Sevilla. (4) Instituto de Investigaciones Biomédicas Alberto Sols. Madrid.
El Síndrome de Allan-Herndon-Dudley (SAHD) es un grave retraso psicomotor que asocia paraparesia espástica, epilepsia y finalmente caquexia que conduce a una muerte temprana. Se debe a mutaciones del gen MCT8, localizado en el cromosoma X. MCT8 transporta las hormonas tiroideas T3 y T4 al interior de muchos órganos, siendo especialmente importante en el cerebro. Su perfil tiroideo es característico: T3L muy elevada, T4L baja y TSH levemente incrementada. Los únicos intentos terapéuticos en el SAHD (PTU+L-T4 y DITPA) no han conseguido modificar el deterioro neurológico progresivo de la enfermedad.
Objetivo: Evaluación de la eficacia del tratamiento compasivo con TRIAC en un niño con SAHD. Comparación de la actividad biológica del TRIAC con la T3 en cultivos de fibroblastos obtenidos de la piel del paciente.
Paciente y Métodos: Paciente de 6 meses con hipotonía axial, retraso psicomotor, hipotiroidismo (TSH 7.7 µU/mL, T4L 0.48 ng/dL) con T3L elevada (8.86 pg/mL). PCR y secuenciación de MCT8 y estudio CGH (Thyroarray©). Tratamiento con dosis crecientes de TRIAC (entre 10 y 40 µg/kg/día), seguimiento del neurodesarrollo con el test de Brunet-Lezine. Obtención de fibroblastos del paciente mediante biopsia cutánea, cultivo y tratamiento in vitro con 1 y 10 nM de T3 o TRIAC, cuantificando por RT-PCR la expresión de genes diana de T3 (RCNA2, HR, BTEB1).
Resultados: Deleción de novo en MCT8 de 25.04 Kb. In vitro, TRIAC tiene la misma actividad que T3 en la expresión de genes diana. El tratamiento compasivo con TRIAC desde los 9-16 meses de vida, corrigió los niveles de T3L y TSH (4.1 pg/mL y 1.2 mU/L) y disminuyó la T4L (0.3 ng/dL). Tras 7 meses de tratamiento, el desarrollo motor experimentó un incremento significativo (del 18% al 40%), y el área social y lenguaje se mantienen estables en torno al 72%.
Conclusiones: El tratamiento con TRIAC, administrado por primera vez en un paciente con SAHD, revierte el hipertiroidismo y la hipertirotropinemia. La hipotiroxinemia necesita de adición de levotiroxina. En corto plazo, TRIAC enlentece el deterioro neurológico característico del SAHD en nuestro paciente, sugiriendo que atraviesa la barrera hematoencefálica y es funcional a nivel neuronal.
Suprarrenales
O4/d3-025 CORRELACIÓN GENOTIPO-FENOTIPO EN FORMAS NO CLÁSICAS DE HSC POR DÉFICIT DE 21-HIDROXILASA EN UN HOSPITAL TERCIARIO AC. Barreda Bonis 1, J. Rodríguez Contreras 1, L. Salamanca Fresno 1, J. Guerrero-Fernández 1, B. Ezquieta Zubicaray 2, I. González Casado 1.
(1)S. Endocrinología Infantil. Hospital la Paz. Madrid. (2) Laboratorio de Diagnóstico Molecular. S. Bioquímica. Hospital Materno Infantil Gregorio Marañón. Madrid
Introducción: Aunque para determinadas mutaciones de CYP21B existe una relación fenotípica clara, en especial con las formas clásicas, en los casos de formas no clásicas (HSC-NC) no siempre es fácil concordar la clínica con el genotipo encontrado.
Material y Métodos/objetivos: Revisión retrospectiva de los casos afectos de HSC-NC en seguimiento por nuestro centro entre los años 1999-2013; definidas por pico de 17OHprogesterona superior a 11.95 ng/dl tras test de ACTH. En función de los resultados genéticos obtenidos hasta el momento los pacientes se clasificarán como formas con mutación leve en homocigosis, leve-grave (formas afectas), grave-normal, leve-normal y normal-normal (portadores con hiperandrogenismo), especificando las mutaciones obtenidas para cada subtipo.
Se recogen como variables clínicas: sexo, edad de inicio de síntomas, valores de 17OH progesterona basales y pico en el test de ACTH, talla adulta, diferencia con respecto a talla genética, inicio de pubertad y crecimiento durante la misma, y desarrollo evolutivo de sobrepeso, hiperandrogenismo e hiperinsulinismo. Se analizará si existen diferencias entre las mismas en función de la genética.
Resultados: 58 pacientes (V/M: 20/38). Concordancia con la genética obtenida en 40/58 (69%) (formas leves en homocigosis y graves/leves en heterocigosis compuesta). 55 casos recibieron tratamiento con hidrocortisona (dosis media 8±2,9 mg/m2; duración de 5,7±3,5 años). Los datos se recogen en la Tabla adjunta.
Existen diferencias estadísticamente significativas en edad de presentación más precoz y en el ratio EO/EC, a favor de las formas con alelos grave/leve, así como en los valores de 17OHprogesterona (basal-pico). No se encontraron diferencias respecto a la talla genética y en el crecimiento durante la pubertad.
Conclusiones: La concordancia clínico-genética en nuestra serie es algo inferior a la obtenida por otros autores (69 vs 72%). Parece que las formas con alelos grave/leve presentan una clínica más intensa al diagnóstico, respecto a edad de presentación, ratio EO/EC y niveles de 17 OH progesterona. Nuestra serie no encontró diferencias significativas en talla adulta con respecto a talla genética, ni tampoco en el crecimiento puberal, estando el 95% de nuestros casos tratados con hidrocortisona. Es preciso tipificar correctamente ambos alelos y hacer un seguimiento estrecho a portadores con hiperandrogenismo.
O4/d3-026 RESPUESTA DEL CORTISOL A LA PRUEBA DE ACTH EN PACIENTES CON HIPERPLASIA SUPRARRENAL CONGÉNITA FORMA NO CLÁSICA YP. Oyakawa Barcelli, B. Roldán Martín, M. Martín Frías, R. Barrio Castellanos, D. Alonso Martín, M. Alonso Blanco.
Hospital Universitario Ramón y Cajal. Servicio de Pediatría. Madrid.
Introducción: Diferentes estudios sugieren, que aproximadamente, un tercio de los pacientes con Hiperplasia Suprarrenal Congénita Forma No Clásica (HSCNC) presentan una respuesta subóptima del cortisol a la prueba de ACTH. Las últimas guías clínicas recomiendan el tratamiento con glucocorticoides (GC) de este subgrupo de pacientes en situaciones de estrés aún cuando no estén recibiendo tratamiento sustitutivo con GC.
Objetivo: Analizar la respuesta del cortisol al ACTH en pacientes con HSCNC y valorar si es necesario el tratamiento con GC a dosis de estrés en pacientes con respuesta insuficiente del cortisol.
Materiales y Métodos: Estudio descriptivo y retrospectivo de 46 pacientes con HSCNC diagnosticados y en seguimiento en nuestra Unidad. Valoramos en los test de ACTH (250 mcg) realizados, la respuesta del cortisol y consideramos normal toda respuesta > 18 mcg/dl. Revisamos que pacientes recibían tratamiento con GC y la pauta de actuación ante situaciones de estrés.
Resultados: De los 46 pacientes, a dos de ellos no se les realizó el test de ACTH por tener una 17OHP basal diagnóstica. De los 44 pacientes a los que se le realizó la prueba de ACTH, solo se determinó el cortisol en 34 pacientes, de ellos, 9 (26%) presentaron respuesta <18mcg/dl a los 60 minutos (Tabla). De estos 9 pacientes, 8 iniciaron tratamiento con GC por avance madurativo o signos de hiperandrogenismo. De acuerdo con guías anteriores, los pacientes no fueron informados sobre la necesidad de tratamiento con GC en situaciones de estrés y no consta que presentaran mala tolerancia ante cuadros infecciosos.
Conclusiones: 1. Un 26% de nuestros pacientes con HSCNC presentaron una respuesta subóptima del cortisol a la prueba de ACTH. 2. Ninguno de ellos presentó síntomas/signos compatibles con insuficiencia suprarrenal ante enfermedad o estrés importante. 3. Nuestros resultados no sostienen la necesidad de aportar GC en situaciones de estrés en estos pacientes.
O4/d3-027 ÍNDROME DE CUSHING EN NIÑOS Y ADOLESCENTES: ESTUDIO RETROSPECTIVO DE SU PRESENTACIÓN, DIAGNÓSTICO, MANEJO Y EVOLUCIÓN EN UN HOSPITAL TERCIARIO DE LONDRES M. Güemes Hidalgo1, P. Murray 2, M.T. Dattani 3.
(1)Departamento de Endocrinología. Great Ormond Street Hospital for Children (Londres, Reino Unido). (2) Paediatrics and Adolescent Endocrinology Department, University College London Hospital. (Londres, Reino Unido). (3) Endocrinology Dept, Great Ormond Street Hospital. Paediatrics Endocrinology Dept, University College London Hospital. Developmental Endocrinology Research Group, Clinical and Molecular Genetics Unit, Institute of Child Health. (Londres, Reino Unido)
Introducción: El síndrome de Cushing (SC) pediátrico es una patología infrecuente pero con significativa morbi-mortalidad dado su desafío diagnóstico al tener una presentación clínica heterogénea y a menudo resultados inconcluyentes en las pruebas.
Objetivo: Describir la experiencia de un centro pediátrico terciario de Londres en el diagnóstico, tratamiento y seguimiento de pacientes con SC.
Material y Métodos: Se estudiaron retrospectivamente las características clínicas, bioquímicas y radiológicas de los pacientes referidos a este centro con diagnóstico de SC entre los años 1983 y 2013. Resultados: Se identificaron 30 pacientes (14 mujeres). La mediana en edad de presentación fue 8,9 años (rango 0,2 - 15,5) y el retraso entre inicio de sintomatología y diagnóstico fue 1,0 año (rango 0,04 - 6). Las manifestaciones más comunes fueron ganancia ponderal (23/30), hirsutismo (17/30) y acné (15/30). La mediana del IMC en el momento de presentación fue +2,27 DE (rango -6,5 - +4,6); 15 pacientes tenían un IMC >2,0 DE mientras que 3 pacientes con hipertensión tenían un IMC <0 DE. La hipertensión estaba presente en 8/30.
16 pacientes tenían un adenoma hipofisario secretor de ACTH, 11 hipercortisolemia de origen adrenal, 2 secreción ectópica de ACTH y en 1 caso la etiología permanece desconocida. La Tabla recoge los resultados de las pruebas realizadas.
Se obtuvo confirmación genética en un carcinoma adrenal con mutación de línea germinal en p53, y en un SC congénito con mutación heterozigota en GNAS1.
Todos los pacientes fueron operados. 8 recibieron metirapone pero tuvo que ser suspendido en 4 debido a los efectos secundarios. Todos necesitaron sustitución glucocorticoidea tras la operación, siendo esta interrumpida en 12 pacientes tras una duración mediana de 1,1 años (rango 0,5-2,1). La mediana de IMC 2 años post tratamiento fue 1,1 DE (rango -1,0 - +3.9).
Las complicaciones incluyeron deficiencias hormonales (17/30; transitorias y/o persistentes), resección incompleta (4/30), recaída (5/30), segunda neoplasia (1/30) y fallecimiento (1/30).
Conclusiones: Un número pequeño de pacientes hipertensos con SC se manifiestan atípicamente con IMC DE bajo. A pesar de que todos los pacientes necesitarán sustitución glucocorticoidea, casi la mitad serán capaces de interrumpirla 1 año tras el tratamiento del SC.
Gónadas
O4/d3-028 UTILIDAD DE LOS DISTINTOS TIEMPOS DE EXTRACCIÓN Y PICOS HORMONALES EN EL TEST DE LHRH PARA EL DIAGNÓSTICO DE PUBERTAD J. Lazaro Ramos, J. Posadilla, P. González, E. Serrano, M. Matín, P. Prieto Matos.
Hospital Universitario de Salamanca / Servicio de Pediatría. Salamanca.
Introducción: El diagnóstico de pubertad se basa en unos criterios clínicos y analíticos siendo el test de LHRH la prueba de oro para el diagnóstico de pubertad. En el momento actual existe controversia en los tiempos de extracción y los puntos de corte.
Objetivo: Analizar el momento y la importancia del pico hormonal en el test y su relación con la clínica.
Pacientes y Métodos: Estudio observacional, retrospectivo de 31 pacientes con sospecha de pubertad, en los que se realiza test LHRH. Definimos como pubertad un pico de LH mayor de 7 U/l ó pico superior a 3 U/l junto a datos clínicos y radiológicos (testes mayores de 6 ml; Tanner III y ecografía puberal; Tanner III y adelanto de la edad ósea más de 2 años). Se analizan los datos con el paquete estadístico SPSS17.
Resultados: Se revisan 31 test de pacientes con sospecha de pubertad. Un 84% son puberales (23 con un pico mayor de 7 U/l). El 92.3% son niñas (55.5% con ecografía puberal). El 69,2% han nacido en España. El 65,4% presentan Tanner mayor de II. El adelanto de la edad ósea es 1.85±0.98 años.
En un 54% se alcanza el pico máximo de LH a los 40 minutos.
El 42,3% presenta valores basales de LH mayores de 3 U/L y el 21,7% mayores de 7 U/L. A los 20 minutos el 100% tienen valores de LH mayores de 3 U/L y el 88,4% mayores de 7 U/L.
El 30,7% de los pacientes presentan criterios de pubertad al inicio del test y el 100% de los pacientes presentan los criterios definidos de pubertad en el minuto 20.
Conclusiones: Los valores basales de LH son suficientes para el diagnóstico de pubertad en un número significativo de pacientes. Todos los pacientes cumplen criterios de pubertad a los 20 minutos del test.
Hipotálamo-Hipófisis
O4/d3-029 Pubertad precoz central en mujeres: Hallazgos clínicos, analíticos y radiológicos M. Cemeli Cano, M. López Úbeda, A. De Arriba Muñoz, B. Martínez Ganuza, M. Ferrer Lozano, J.I. Labarta Aizpún.
Hospital Universitario Miguel Servet (Servicio de Pediatria). Zaragoza.
Introducción: La realización de resonancia magnética (RM) en mujeres con pubertad precoz (PP) está recomendada en menores de 6 años según la bibliografía reciente sin embargo, no se han descrito variables predictoras lo suficientemente seguras como para seleccionar a qué tipo de niñas se les debe realizar.
Objetivo: Analizar los antecedentes, hallazgos analíticos y radiológicos en una cohorte de mujeres con PP, teniendo en cuenta diferentes variables que podrían influir en su evolución y predicción de la presencia de hallazgos patológicos en la RM.
Material y Métodos: Revisión de historias de 123 mujeres con diagnostico de PP central, de las cuales se realizó RM al 62,6%. Se compararon: antecedentes personales, medidas antropométricas, edad ósea, niveles de estradiol, LH, FSH, hallazgos radiológicos y necesidad de tratamiento.
Resultados: Los resultados se presentan en la Tabla adjunta. Se realizó RM a 74 pacientes, 16,2% con hallazgos radiológicos. Los principales diagnósticos fueron: 5 tumores (2 microadenomas hipofisarios y 3 hamartomas), 2 quistes (glándula pineal y 3º ventrícu-
Conclusiones: La PP central representa generalmente una entidad benigna, sin embargo el 16.2% de nuestras mujeres a las que se realizó RM, presentaron un hallazgo patológico en la RM. Un estadio puberal avanzado al diagnóstico es el único factor asociado con anomalía orgánica en la neuroimagen.
Genética
O4/d3-030 ESTUDIO SOBRE CALIDAD DE VIDA Y SITUACIÓN CLÍNICA EN LA EDAD ADULTA EN MUJERES CON SÍNDROME DE TURNER DESPUÉS DE LA TRANSICIÓN: ESTUDIO PRELIMINAR S. Congost Marín1, P Sanjuan Casamayor2, A de Arriba Muñoz2, M Ferrer Lozano2, E Mayayo Dehesa2,
(1)Hospital de Barbastro/Pediatría. (2) Hospital Maternoinfantil Miguel Sevet, Zaragoza.
Introduccion: Las mujeres con síndrome de Turner (ST) presentan mayor morbimortalidad que la población general en la edad adulta y ello repercute sobre su calidad de vida.
Objetivo: Evaluar el estado de salud y calidad de vida de mujeres adultas con ST.
Material y metodos: Estudio descriptivo observacional mediante revisión de historias clínicas, entrevista personal y realización del cuestionario de calidad de vida SF-36. Estadística descriptiva y comparación de medias (p<0.05).
Resultados: Muestra de 18 mujeres con edad media de 35,66 años (rango 21,09-57,1) controladas previamente en nuestra Unidad; cariotipo más frecuente es 45 X0 en el 50%, seguido de 46 XXi (11%), 46 XXq- (5%), y tres casos de mosaicos (45X0/45Xi/46XX, 45X0/46XX, 45X0/46XXi); edad media al diagnóstico de 6,2 años. La talla media actual es de 148.8±6,4 cm; 72% recibieron tratamiento con GH (talla media de 149.07±7,3 cm) y el resto no (talla media de 148.02±1.2cm). El 100% presentan comorbilidad asociada, siendo el eje ORL (hipoacusia) y alteraciones metabólicas (obesidad, HTA,…) las mas frecuentes (ver Tabla 1). A lo largo del último año el 88,9% han acudido a revisión médica siendo el Servicio de Endocrinología el más frecuentemente visitado (61%), seguido de Otorrinolaringología y Ginecología (27.8%) y Dermatología (16.7%) . El grupo Turner ha conseguido mejores resultados en todos los ítems de función física y mental que el grupo control, sin hallar diferencias estadísticamente significativas (p>0.05). Al agruparlas por grupos de edad tampoco se ha observado diferencias significativas aunque, al igual que en los datos poblacionales, se obtienen peores cifras en el cuestionario conforme aumenta la edad de las encuestadas. Respecto a la evolución de la salud en el tiempo el 61% de las encuestadas creen que su salud es similar al año previo y el 16% que ha empeorado.
Conclusiones: Las pacientes con ST presentan mayor morbilidad que la población general lo que no parece afectar a su calidad de vida. Al avanzar la edad de las pacientes con ST su calidad de vida se ve más afectada que en la población de control y ello ocurre a expensas del componente físico.
O4/d3-031 EVALUACIÓN DEL SEGUIMIENTO ACTUAL DE LOS PACIENTES CON SÍNDROME DE PRADER-WILLI EN NUESTRO MEDIO L. Calvo Naharro, J. Pérez Sánchez, L. Sánchez Barnés, E. Gabau Vila, A. Caixàs Pedregosa, R. Corripio Collado.
Corporació Sanitària i Universitària Parc Taulí. Sabadell. Introducción: El síndrome de Prader-Willi SPW es una enfermedad genética por falta de expresión del gen paterno 15q11-q13. El diagnóstico precoz, atención temprana, seguimiento multidisciplinar y administración de GH han mejorado su calidad de vida cambiando la evolución natural de la enfermedad.
Actualmente no existen guías en España para el adecuado manejo y seguimiento de estos pacientes.
Objetivo: Determinar la situación actual del manejo de pacientes con SPW en nuestro medio y objetivar puntos mejorables en su control y seguimiento.
Material y Métodos: Desarrollamos una encuesta basada en guías clínicas actualizadas recogiendo datos demográficos, de seguimiento y tratamiento considerados como óptimos.
Propusimos la participación a familiares de pacientes mediante un formulario al cual se accedía a través de un enlace web proporcionado por la asociación de familias de SPW. Obtuvimos la aprobación por el CEIC de nuestro centro.
Resultados: Recogimos 27 pacientes (15 varones, 12 mujeres). Edad actual media 9.8±7.2 años (5 adultos) y al diagnóstico 15.3±24.8 meses, 50% diagnosticados antes de los 6 meses. El 90% presentan delección paterna o disomía uniparental materna. IMC actual: +0.86±2.8 SD (3 adultos tienen IMC >2SD). Diez pacientes visitados en centro de referencia además de Hospital de zona.
Seguimientos realizados (Tabla anexa). Del 50% de los seguidos en nutrición, el inicio es posterior al año de vida. Mediana de inicio en Estimulación Precoz: 4 meses de edad. Se realizó seguimiento puberal en 9/10 adolescentes, dos con tratamiento hormonal sustitutivo. Edad de inicio de GH: 4.1±2,6 años, en el 30% inicio antes de los 2 años, en 5 se suspendió (media de finalización: 12.3±2.8 años). El 53% presentó patología del sueño.
Comentarios: -El seguimiento por nutricionista, soporte psicológico y neumólogo parece ser insuficiente a pesar de lo importante que es para estos pacientes y familiares. -El control nutricional se inicia tarde, paradójicamente la prevalencia de obesidad es baja. -Hay tendencia a finalizar el tratamiento con GH prematuramente. -Debemos esforzarnos para elaborar guías de adecuado manejo y seguimiento haciendo hincapié en medidas nutricionales, neumológicas, soporte familiar y sobre todo el tratamiento con GH precoz ya que modifica la evolución natural de la enfermedad.
Displasias óseas
O4/d3-032 IMPLEMENTACIÓN DE UN ENSAYO DE SECUENCIACIÓN MASIVA PARA EL DIAGNÓSTICO DE LAS DISPLASIAS ESQUELÉTICAS (PROYECTO ENDOSCREEN) KE. Heath 1, E Vallespín 1, A del Pozo 1, E Mansilla 1, VEF Montaño 1, J Nevado 1, J. Carlos Silla 1, K. Ibañez 1, L. Sentchordi 2, C.I. Rivera Pedroza1, A.C Barreda Bonis 3, F. Santos 1, S. García-Miñaur 1, I. González Casado 3, Á. Campos Barros, P. Lapunzina 1.
(1)Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, UAM, IdiPAZ y CIBERER, ISCIII, Madrid. (2) Servicio de Endocrinología Pediátrica, Hospital Universitario Infanta Leonor, Madrid, (3) Servicio de Endocrinología, Hospital Universitario La Paz, UAM, Madrid
Introducción: Se conocen 456 tipos de displasias esqueléticas (DESQ) clasificados en 40 grupos distintos. Su correcto diagnóstico es dificil, debido a su gran variabilidad fenotípica y también genotípica, con más de 226 genes asociados. El incremento de la demanda del diagnóstico molecular de las DESQ, incluso a un nivel prenatal, el bajo nivel de identificación del defecto molecular causativo en muchas de ellas y el desconocimiento del gen implicado en un 24% de las mismas, ha puesto de manifiesto la necesidad de mejorar su diagnóstico molecular mediante la técnica de secuenciación masiva (Next generation sequencing, NGS) para abordar el análisis simultáneo de todos los genes implicados en un único ensayo.
Objetivo: Evaluar y comparar dos paneles de NGS para el diagnóstico molecular de las DESQ.
Métodos: Diseño 1: Captura mediante la tecnología de Haloplex (Agilent Technologies) y secuenciación en una plataforma MiSeq (Illumina) de un panel de 248 DESQ genes en 33 muestras con mutaciones conocidas y 8 sin mutación conocida. Diseño 2: Captura y secuenciación de un nuevo panel comercial de 4.813 genes asociados a un fenotipo clínico (TruSight-One, Illumina) en 5 muestras sin defecto molecular conocido. Las nuevas mutaciones identificadas y hallazgos fueron confirmados mediante secuenciación Sanger.
Resultados y conclusiones: Diseño 1: Aunque la cobertura de los genes ha sido inferior a la esperada, el protocolo de NGS implementado detectó la mayoría de las variantes de nucleótido único así como las que alteran la dosis génica (CNV), mostrando sin embargo, una sensibilidad limitada para detectar indels, así como para capturar secuencias con regiones repetitivas, como la región PAR1 del gen SHOX. Diseño 2: Actualmente, estamos analizando los datos generados. La aplicación de NGS nos ha permitido identificar nuevas mutaciones en algunos casos de DESQ sin mutación conocida, tales como dos mutaciones en WISP3 y una mutación en NPR2. Para la implementación de NGS en el diagnóstico clínico es fundamental el consejo genético de los pacientes, así como el uso de un consentimiento informado apto para esta tecnología en el que se informe al paciente sobre la posible obtención de hallazgos incidentales y su posible actitud ante los mismos. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites