
| Rev Esp Endocrinol Pediatr 2011;2 Suppl(1):145-156 | Doi. 10.3266/RevEspEndocrinologPediatr.pre2011.May.59 |
| Tiroides |
| Sent for review: 2 May. 2011 | Accepted: 3 May. 2011 | Published: 3 May. 2011 |
| Congreso SEEP |
| Correspondence: Congreso SEEP E-mail: seep@seep |
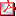 Tabla - P1d2-3-101_012 Tabla - P1d2-3-101_012 |
| Tabla - P1d2-3-103_012_ectopias |
| Tabla - P1d2-3-108_012_ca tiroides |
| Tabla - P1d2-3-110_012_Hemiagenesia tiroidea |
|
|
P1/d2-3-094 TUMOR DE WILMS EN LA INFANCIA COMO RIESGO POTENCIAL PARA UN CÁNCER SECUNDARIO TIROIDEO. J. Nieto Cuartero, L. Madero López, D. Azorín Cuadrillero, E. García Esparza, JC. Ollero Caprani, JM. Ollero Fresno.
Hospital Infantil Universitario Niño Jesús, Madrid.
Introducción: Aunque la mayoría de los segundos tumores malignos suelen desarrollarse en los tumores de Wilms después de la exposición a la radioterapia, no siempre sucede así ya que pacientes tratados sólo con quimioterapia, han desarrollado cánceres de tiroides secundarios y más si existe como en nuestro caso un riesgo sobreañadido ya que la madre del paciente que presentamos ha sido operada de un carcinoma papilar tiroideo. En la revisión del St Jude Children´s Hospital entre febrero de 1962 y febrero del 2002, se encontró un caso de tumor de Wilms asociado a carcinoma tiroideo.
Material y Métodos: Presentamos un varón que a los 3 años de edad, es diagnosticado de tumor de Wilms extrarrenal trifásico con áreas anaplásicas en enero de 1999. Fue intervenido con resección de la tumoración, aparentemente completa e inició tratamiento quimioterápico según protocolo de la SIOP 93, estadio III con buena tolerancia. Posteriormente reingresó para aféresis y trasplante autólogo de progenitores hematopoyéticos de sangre periférica previa movilización con G-CSF a 24 mg/kg/día repartido en dos dosis durante cuatro días. A la edad de 14 años y 5 meses presenta una talla de 1,71 cm y peso 56,7 Kg, edad ósea de 14 años Tanner III, en la analítica se aprecia positividad de los anticuerpos antitiroideos, por lo que se realiza una ecografía tiroidea que demuestra dos formaciones nodulares en el lóbulo tiroideo izquierdo de contenido coloideo de 18x17 mm en vista de lo cual se realiza PAAF, que no muestra por el momento células neoplásicas malignas y sí dos nódulos quísticos coloideos. Se advierte a la madre que tendrá que tener un seguimiento de por vida, por el riesgo del 15% de esos nódulos coloideos de que puedan ser en el futuro carcinoma papilar (y más teniendo en cuenta que la madre ha sido operada de un carcinoma papilar con tiroidectomía total).
Resultados: Nuestro caso muestra el riesgo de la aparición de posibles carcinomas tiroideos en pacientes operados y no radiados de tumor de Wilms
Conclusiones: Todo tumor de Wilms tiene que ser vigilado, por el riesgo de presentar un carcinoma tiroideo en el futuro.
P1/d2-3-095 CARCINOMA FOLICULAR DE TIROIDES EN NIÑA DE 5 AÑOS SIN EXPOSICIÓN EVIDENTE A RADIACIÓN. A. Mingorance Delgado, N. Gilabert Martínez, B. Castillo Gómez.
Unidad de Endocrinología Infantil. Servicio de Pediatría. Hospital General Universitario de Alicante.
Presentamos el caso de una niña de 5 años con nódulo tiroideo, de 1 semana de evolución. Disfonía progresiva, sin fiebre. No antecedentes de irradiación, salvo radiografías de cavum por hipertrofia adenoidea, intervenida un mes antes del diagnóstico.
Exploración física: Aumento de volumen del lóbulo tiroideo derecho (LTD) de consistencia dura y aproximadamente 3 cm de diámetro. No adenopatías patológicas.
Exploraciones complementarias: Función tiroidea normal con anticuerpos negativos. Ecografía tiroidea: LTD con 2 imágenes nodulares heterogéneas.
Gammagrafía: Nódulo hipocaptante que ocupa el tercio medio e inferior del LTD.
Por la edad del paciente y la progresión rápida se realizó hemitiroidectomía derecha con anatomía patológica diagnóstica de carcinoma folicular limitado a tiroides, rodeado por una gruesa cápsula focalmente infiltrada e invasión de vasos capsulares. Se completa tiroidectomía, con preservación de una de las glándulas paratiroides y se administra dosis ablativa de I-131. Controles posteriores de tiroglobulina negativos con cifras de Ca y PTH normales que permiten retirar parcialmente el tratamiento con calcitriol y calcio. Estudio de extensión con gammagrafía de rastreo y test Thyrogen negativos.
Se ha seguido anualmente con controles de Tiroglobulina y anticuerpos antitiroglobulina y los necesarios para ajuste de dosis supresora de Levotiroxina. En la evolución presenta episodios cada vez más frecuentes de tetania sin respuesta de PTH, precisando volver a tratar con suplementos de calcio y vitamina D3.
Conclusiones: La presentación en niños pequeños de nódulos tiroideos con ausencia de clínica tiroidea nos ha de hacer pensar en una posible etiología maligna. Los carcinomas foliculares de tiroides son muy infrecuentes en menores de 10 años. En ninguno de los casos comunicados la edad ha sido inferior a 6 años. La preservación de una de las glándulas paratiroides no ha evitado la clínica de hipocalcemia a medio plazo. La retirada de tratamiento sustitutivo para evaluación de recurrencia del carcinoma, puede resultar problemática. Algunos protocolos recomiendan seguir los carcinomas diferenciados de tiroides con controles de tiroglobulina y sus anticuerpos.
P1/d2-3-096 UNA ASOCIACIÓN NO SIEMPRE RECORDADA: ENFERMEDAD TIROIDEA AUTOINMUNE Y GASTRITIS AUTOINMUNE. M. Bonet Alcaina, S. Ortigosa Gómez, P. Guevara Carrasco, C. Alvarez Urturi.
Parc de Salut Mar, Barcelona.
Introducción: La enfermedad tiroidea autoinmune (ETAI) que incluye la tiroiditis linfocitaria crónica y la enfermedad de Graves es un trastorno autoinmune órgano específico definido por la infiltración linfocítica del tiroides y presencia de anticuerpos antitiroideos. Se asocia frecuentemente a otras enfermedades autoinmunes como diabetes mellitus tipo1, celiaquía y gastritis autoinmune (GA).
La GA es una gastritis crónica caracterizada por pérdida de las glándulas oxínticas, responsables de la secreción de ácido clorhídrico y factor intrínseco. La hipoclorhidria causa hipergastrinemia y disminución de pepsinógeno I. La GA va asociada a anemia ferropénica y perniciosa e incrementa el riesgo de cáncer gástrico y tumores carcinoides. Los anticuerpos contra las células parietales gástricas (ACPG) son el principal marcador inmunológico de GA.
La prevalencia de GA en pacientes con ETAI es de 3 a 5 veces superior a la población general (2%).
Caso clínico: Adolescente de 18 años diagnosticada desde hace 4 años de enfermedad de Graves tratada con metimazol siendo el cumplimiento terapéutico irregular. Granuloma anular. Anemia ferropénica con buena respuesta al tratamiento con hierro.
En el estudio de GA, se detecta ACPG positivos, hipergastrinemia, pepsinógeno I disminuido y serología positiva para Helicobacter pylori. Se practica endoscopia digestiva alta con toma de biopsia en al que se observa presencia de gastritis atrófica e hiperplasia neuroendocrina micronodulillar con escasos gérmenes compatibles con Helicobacter pylori. Tras tratamiento erradicador de este gérmen con resultado positivo persiste hipergastrinemia. Se solicita cromogranina A que muestra valores elevados y gammagrafía con octreótido que no evidencia proceso tumoral neuroendocrino.
Conclusiones: - Dada la alta prevalencia de asociación entre ETAI y GA, aún en ausencia de síntomas, se recomienda la investigación de ACPG al diagnóstico de ETAI y en caso de negatividad repetir el estudio cada dos años. - En presencia de ACPG positivos se aconseja determinación anual de gastrina. La hipergastrinemia constituye un indicador fiable de GA y establece la indicación de endoscopia digestiva alta. - El diagnóstico de la GA es importante para tratar sus posibles complicaciones. - La infección por Helicobacter pylori podría estar implicada en la inducción de GA, por lo que se recomienda su investigación y tratamiento.
P1/d2-3-097 HIPERTIROIDISMO NEONATAL. A PROPÓSITO DE UN CASO. P.M. Lalaguna Mallada, S. Conde Barreiro, M. Bustillo Alonso, MªJ. Calvo Aguilar.
Hospital de Barbastro, Huesca.
Se presenta un caso de hipertiroidismo neonatal por paso transplacentario de anticuerpos estimulantes del receptor tiroideo de tirotropina (TSH-Rs), en un recién nacido hijo de una madre con enfermedad de Graves
Caso clínico: Recién nacido varón. Parto vaginal eutócico a las 34,3 semanas de gestación. Apgar 9/10. Impregnación meconial y exoftalmos bilateral. P: 1975 gr. L: 44,7 cm. PC: 30,5 cm. Madre afecta de enfermedad de Graves, tratamiento con propiltiouracilo (100 mg/día) desde la semana 11 de gestación (T4L: 6,4 ng/dL, TSH: <0,05 mcU/ml, TSH-Rs: 97,2 U/L), resto de embarazo eutiroidea. Al segundo día de vida, el niño presenta taquicardia sinusal con extrasístoles ventriculares, tensión arterial elevada, irritabilidad, dificultad para la alimentación con pérdida excesiva de peso y exoftalmos más llamativo. Analítica TSH: 0,011 mcU/ml. T4L: 7,47 ng/dL y TSH-Rs: 12,5 U/L. Ecografía tiroidea: tiroides aumentado de tamaño. Se administra propiltiouracilo a 5 mg/kg/día, solución de lugol (2 gotas/8 horas, un total de 8 dosis) y propanolol a 1 mg/kg/día (5 días). Precisa colocación de sonda nasogástrica para alimentación. Tras 48 horas de tratamiento control TSH: 0,009 mcu/ml. T4L: 1,86 ng/dL. A los 14 días se añade levotiroxina a 5 mcg/Kg por estado hipotiroideo. Presenta curva ponderal ascendente. Alta a los 18 días con peso 2.200 gr. Posteriormente se ajustan dosis según controles analíticos. El niño se mantiene eutiroideo y asintomático. Se reduce la dosis de propiltiouracilo hasta suspenderla en el cuarto mes cuando se negativizan los anticuerpos TSH-Rs. La tiroxina se mantiene hasta los 6 meses por persistencia de TSH suprimida. Tras la suspensión del tratamiento, presenta función tiroidea normal.
Conclusiones: - La infrecuencia de este cuadro, y su potencial gravedad, hacen necesario un riguroso control de los hijos de madre afecta de enfermedad de Graves. - La sintomatología y la determinación de la función tiroidea permite el diagnóstico y la instauración rápida del tratamiento para controlar la situación metabólica. - Es un trastorno transitorio, pero requiere un estrecho seguimiento para ajustar el tratamiento y garantizar una correcta función tiroidea hasta la negativización de los anticuerpos.
P1/d2-3-098 ENFERMEDAD DE GRAVES EN EDAD ESCOLAR QUE DEBUTA CON EXOFTALMOS. M.A. Santos Mata, F.J. Macías López, L. Muñoz Núñez, S. Rodríguez López, J. Ortiz Tardío.
Hospital SAS Jerez de la Frontera, Jerez de la Frontera.
Introducción: La enfermedad de Graves es la causa más frecuente de hipertiroidismo en la población pediátrica, si bien su prevalencia es baja 0,02%, siendo muy infrecuente en edades inferiores a los 10 años.
Caso clínico: Niña de 9 años que presenta de forma brusca dolor ocular, destellos con la luz, e imposibilidad para cerrar los párpados. No pérdida de peso, leve sudoración de forma ocasional.
Antecedentes personales: Embarazo controlado. Parto espontáneo, eutócico. Auxología neonatal: normal.
Examen Físico: Peso: 30 kg (P:50), Talla: 138cm (P:75). Proptosis ocular bilateral, edema e hiperemia conjuntival y palpebral, con retracción palpebral y restricción de movimientos oculares. No bocio. AC: no soplo. FC: 110 lpm, TA: 108/50 mmhg. No masas no megalias. Tanner: 1. Exámenes complementarios: Hemograma: anemia normocrómica normocítica. Perfil básico, renal, hepático, lipídico: normales. TSH: 0,005 mcUI/ml, T4 libre: 3,7 ng/ml, T3 libre: 11,8 pg/ml, Ac antiitroglobulina: 1890UI/l, Ac. antiperoxidasa>600 UI/ml, TSI: 15U/l, Tiroglobulina: 2,4 ng/ml.ANA: negativo, Ac Anticelulas parietales: negativos. Somatomedina C: 207 ng/ml, IGFBP·: 3,5 mcg/ml. IGA,IGG,IGM: normales. Serología de gluten: negativa. Ecografía ocular: músculos rectos engrosados con tendón conservado. RNM de órbita: leve engrosamiento músculos rectos. Ecocardiografía: normal. ECG: normal, QT en límites. Holter: Normal. Ecografía tiroidea: tiroides discretamente aumentado de tamaño. Hipervascularización muy llamativa. Importante formación de pseudonódulos por tractos fibróticos. Ligera disminución de la ecogenicidad, itsmo preservado. Gammagrafia tiroidea: bocio difuso hipercaptador.
Evolución: Se inició tratamiento con Carbimazol a 0,5 mg/kg/d así como Prednisona en dosis descendientes 3 semanas, mejorando la sintomatología. Desaparece el exoftalmos a los dos meses de tratamiento. Normalización de la función tiroidea a los 2,5 meses de tratamiento. A los 14 meses se encuentra eutiroidea con negatividad de los anticuerpos.
Comentarios: La enfermedad de Graves es poco frecuente en la edad pediátrica ocurriendo en la mayoría de los casos en niñas entre 10-15 años. No es frecuente el debut con exoftalmos siendo este el único síntoma que presenta la paciente con supresión total de TSH. El interés del caso se centra en su presentación así como en la ecografía y gammagrafía que no son las habituales en esta patología.
P1/d2-3-099 ENFERMEDAD DE GRAVES EN LA EDAD PEDIÁTRICA. S. León Cariñena, M.C. De Mingo Alemany, F. Moreno Macián.
Hospital Infantil la Fe, Valencia.
Introducción: El hipertiroidismo es una patología poco frecuente en la edad pediátrica: la enfermedad de Graves es la causa más frecuente. Se caracteriza por bocio difuso, hipertiroidismo y oftalmopatía.
Los fármacos antitiroideos son el tratamiento inicial de elección, aunque sólo el 25% de los pacientes tras dos años de tratamiento consiguen la remisión y muchos pacientes requieren tratamientos como cirugía o radioyodo.
Material y Métodos: Estudio de cohortes. Análisis descriptivo de 16 pacientes controlados en el Hospital Infantil la Fe de Valencia entre 1999 y 2011.
Las variables cualitativas se describen como %. Las continuas normales (Kolmogorov-Smirnov,
Resultados: 13 mujeres (81.3%) y 3 varones (18.8%) con edad al diagnóstico de 9.04 +/- 3.85 años. El 37.8% asociaban enfermedades autoinmunes y el 68.7% antecedentes familiares: tiroiditis de Hashimoto (31.3%), Diabetes Mellitus tipo 1 y la EG (ambas 25%).
El motivo de consulta más frecuente fue el bocio (62.5%) que presentaba el 100%. El 37.5 % asociaban exoftalmos. El IMC fue de -0.42 SDS +/-0.72 DS. La función tiroidea presentó TSH 0.001 mUI/l (P25 0.001, P75 0.002) y T4 libre 3.50 ng/dl (DS 1.42). El 93.8% tenían anticuerpos antiperoxidasa y el 75% anticuerpos estimuladores del tiroides (TSI).
El tratamiento inicial fue los antitiroideos (15 Carbimazol, 1 Tiamizol) con dosis: inicial 0.45 mg/kg/día (0.10 DS) y máxima 0.56 mg/kg/día (0.20 DS). Tres precisaron betabloqueantes, y al 62.5% se añadió tiroxina tras alcanzar el hipotiroidismo. La duración del tratamiento fue de 2.9 +/- 1.80 años, normalizándose el estado hipertiroideo a los 3.8 meses (P25 2.2, P75 6.7). Tres pacientes presentaron leucopenia, uno un síndrome lupus-like.
5 pacientes (31.3%) alcanzaron la remisión (12 meses eutiroideos tras suspender antitiroideos) y 6 presentaron recaídas (5 no valorables).
Como tratamiento alternativo en los pacientes con recaídas o con efectos secundarios graves se utilizó la cirugía (dos pacientes) y el radioyodo (dos pacientes)
Conclusiones: El alto porcentaje de recaídas con las tionamidas está convirtiendo al radioyodo en primera opción de tratamiento en pacientes adolescentes.
P1/d2-3-100 CÁNCER DE TIROIDES DE RÁPIDA Y AGRESIVA EVOLUCIÓN. M.T. Santos-García Cuéllar(1), J. Sánchez del Pozo (1), L. Garzón Lorenzo (1), J. Cruz Rojo (1), B. García Cuartero(2), M.E. Gallego Gómez (1).
(1) Hospital Doce de Octubre de Madrid, (2) Hospital Severo Ochoa de Madrid.
Introducción: El cáncer de tiroides es una patología infrecuente en la infancia. A esta edad la variedad histológica más frecuente es el papilar. Es fundamental para su sospecha una adecuada exploración del cuello para detectar nódulos tiroideos o metástasis en ganglios cervicales. Caso clínico: Niña de 13 años seguida en consulta de atención primaria por adenopatía laterocervical derecha de 0.5 cm y que presenta crecimiento rápidamente progresivo en 3 meses alcanzando 4 por 6 cm.
Pruebas complementarias: Hemograma, bioquímica y coagulación normales, función tiroidea normal, anticuerpos antitiroglobulina 29.5 UI/ml, antiperoxidasa 253 UI/ml, calcitonina < 2 pg/ml y tiroglobulina 339 ng/ml. Ecografía cervical: masa de 4 por 2 cm en lóbulo tiroideo derecho, que invade músculos pretiroideos y adenopatías laterocervicales, supraclaviculares, paratraqueales y en la salida de grandes vasos. TAC tórax: sin imágenes de metástasis pulmonares. RM: tiroides derecho desestructurado con nódulos en continuación con masa adenopática de 7-8 cm que se extiende hasta el mediastino superior y que se coloca en la salida de grandes vasos y adenopatías laterocervicales y submandibulares múltiples. PET-TC: compatible con neoplasia de tiroides derecha y múltiples adenopatías laterocervicales y mediastínicas.
PAAF: Carcinoma papilar con componente folicular y quístico y ganglios linfáticos congruentes con carcinoma papilar metastásico.
Tratamiento: Tiroidectomía total y vaciamiento cervical bilateral con exéresis de adenopatías mediastínicas. Ablación de restos tiroideos con I131 120 mCi. Levotiroxina 150-175 µg cada 24 horas a días alternos. En el rastreo corporal total post-cirugía se objetivan restos de tejido tiroideo en cara anterior del cuello y acúmulo patológico de material radiactivo en región supraclavicular, laterocervical derecha y 5º y 6º arco costal. Niveles de tiroglobulina tras el tratamiento < 0.2 ng/ml. Actualmente pendiente de nueva ablación con I131 200 mCi.
Conclusión: El motivo de la comunicación de este caso es destacar que la presentación de este tipo de cáncer en niños es más agresiva al diagnóstico que en adultos y en este caso en particular con una agresividad local muy importante. También pueden presentar al diagnóstico metástasis a distancia, sobre todo pulmonares, óseas y cerebrales. Es importante el diagnóstico precoz y un tratamiento rápido y agresivo.
P1/d2-3-101 FUNCIÓN TIROIDEA EN POBLACIÓN INFANTIL OBESA. N. Álvarez Gil, C. Vázquez Ordoñez, M. Martín-Frías, P. Enes Romero, M. Alonso Blanco, R. Barrio Castellanos.
Hospital Ramón y Cajal, Madrid.
Introducción: La obesidad puede alterar los niveles de hormonas tiroideas como consecuencia de una disregulación endocrina entre el eje hipotálamo-hipofisario y el tejido adiposo. Diversos estudios han reportado elevación de la TSH en población obesa.
Objetivos: Investigar, en una población pediátrica obesa, si hay relación entre IMC y otros datos de síndrome metabólico con los niveles de TSH.
Pacientes y Métodos: Estudio prospectivo en 439 pacientes (51,3% varones, 73,6% caucásicos, edad media 11,03±2,7) con obesidad [IMC >2 desviaciones estándar [DE] (Hernández-2004)]. Se valoró: función tiroidea [TSH (vn 0,35-4,95); si alteración, se determinaron T4 y T3 libres, anticuerpos antitiroideos (TPO y TG) y ecografía tiroidea], metabolismo hidrocarbonado [sobrecarga oral de glucosa [SOG] (glucemia, insulinemia)], tensión arterial, perfil lipídico y transaminasas. Definimos síndrome metabólico mediante criterios de Cook modificados. Estudio estadístico realizado con programa SPSS, versión 17.0, mediante pruebas paramétricas. Datos expresados en porcentajes, medias y DE.
Resultados: El 94,5% de los pacientes tenían valores normales de TSH. De los 31 pacientes con TSH elevada, 7 fueron excluidos por presentar anticuerpos antitiroideos positivos; en el resto los niveles de T4 y T3 libres y la ecografía tiroidea fueron normales. No encontramos diferencias significativas al comparar los grupos TSH normal vs elevada en ninguna variable analizada. Encontramos correlación positiva significativa entre niveles de TSH y colesterol y GGT, aunque con coeficiente de correlación <0,2; no encontramos correlación con IMC.
De los 24 pacientes con TSH aumentada sin autoinmunidad tiroidea, 8 recibieron tratamiento con levotiroxina. No detectamos diferencias en la mejoría del IMC al año de seguimiento en función de si habían o no recibido tratamiento.
Conclusiones: No hemos evidenciado relación entre la hipertirotropinemia y el IMC en nuestra población obesa. Es cuestionable si la hipertirotropinemia aislada presente en porcentaje reducido de pacientes obesos tiene alguna repercusión clínica significativa.
P1/d2-3-102 HIPOTIROIDISMO CENTRAL SECUNDARIO A HIPERTIROIDISMO MATERNO. P. Sevilla Ramos, M.J. Alija Merillas, E. Cid París, M.E. Rubio Jiménez, G. Arriola Pereda, J.M. Jiménez Bustos.
Hospital Universitario de Guadalajara.
Introducción: El hipotiroidismo central neonatal es una entidad muy infrecuente en pediatría. Presentamos el caso de una lactante que desarrolló un hipotiroidismo central a los 2 meses de vida, secundario a una enfermedad de Graves materna detectada tras el parto. AP: Embarazo controlado. Cesárea a 34 semanas por RPM, Apgar 9/10 PRN: 2570 (P 75-90); LRN: 45 ( P50-75). AF: Padre sano, talla 170 cm. Madre sana, talla 165 cm
Evolución periodo neonatal: Ingreso al nacimiento por hipoglucemia precoz asintomática. Avisan de screening de metabolopatías por TSH baja, detectándose TSH 0,03 mUI/L y T4L 1,84 ng/dl. H. tiroideas de la madre: TSH indetectable, T4L 2,82, Ac antitiroglobulina 45, Ac antiperoxidasa 75, Ac TSI 13 (N<10). Durante primeros 5 días de vida presenta TA 90/43-78/40, FC 157-130 lpm. Destaca irritabilidad y diarrea.
Seguimiento en consultas: Revisión 15 días: Peso: 2,570 (P3), Longitud: 46 (P3), macroglosia. TSH 0,02 mUI/L, T4L 1,31 ng/dl, T3 1,05 ng/dl, Ac antimicrosomales 71(N 0-5,6). Revisión 2,5 meses : Peso: 4,250 (P3), Longitud: 53 ( Tratamiento levotiroxina: 5,4 mcg/kg/d. Revisión 10 meses: Peso 8,400 (P25), Longitud: 69,5 (P10), inicia gateo. TSH 1,07, T4L 1,09. Tratamiento levotiroxina: 4 mcg/kg/d. Conclusiones: En la enfermedad de Graves materna el paso de Ac TSI transplacentarios afecta el desarrollo del eje hipotálamo-hipofiso-tiroideo (HHT). Esto se traduce en ocasiones a un exceso de estimulación del tiroides fetal dando clínica de hipertiroidismo neonatal transitorio. De forma más infrecuente la exposición a niveles altos de hormonas tiroideas intrauterinas en la enfermedad de Graves materna puede frenar el eje hipotalamo-hipofiso-tiroideo, produciendo un hipotiroidismo en el feto que se mantiene durante un tiempo variable en el RN.
P1d2-3-103 HIPOTIROIDISMO CONGÉNITO PRIMARIO SUBCLÍNICO CON GLÁNDULA TIROIDEA ECTÓPICA. M. Doyle Sánchez (1), M.J. Olmos Jiménez (1), M. Royo Gómez (1), E. Dulín Íñiguez (2), A. Rodríguez Sánchez (1), M.D. Rodríguez Arnao (1).
(1) Unidad de Metabolismo/Endocrinología Pediátrica. Hospital General Universitario Gregorio Marañón. Madrid. (2) Centro de Cribado Neonatal. Hospital General Universitario Gregorio Marañón, Madrid.
Introducción: La utilización de TSH como método de cribado neonatal diagnostica un 24% más de pacientes con hipotiroidismo primario congénito (HC) comparado con la utilización de T4 como parámetro inicial. En España se analiza TSH en muestra de sangre capilar recogida en papel absorbente a las 48 horas de vida mediante inmunofluorescencia (DELFIA), y se realiza T4 total cuando la TSH presenta un valor superior a ≥ 10 µUI/mL.
Sujetos y Métodos: Estudio descriptivo retrospectivo mediante la revisión de historias clínicas de los pacientes diagnosticados de hipotiroidismo congénito ectópico de localización sublingual, con valores de TSH discretamente elevados (10-20 µUI/mL) y T4 total normal.
Resultados: Los datos más relevantes se recogen en la Tabla.
Conclusiones: Los programas de detección precoz de HC deben realizarse en todos en todos los recién nacidos. La determinación de TSH, como método inicial de cribado neonatal es preferible a la determinación de T4. Valores no muy elevados de TSH pueden corresponder a un hipotiroidismo permanente detectado en fase subclínica.
La realización de la gammagrafía tiroidea (*Tc99) al diagnóstico es necesaria para conocer la etiología del HC primario e informar a la familia de la necesidad de mantener de forma permanente el tratamiento.
P1/d2-3-104 ORBITOPATÍA TIROIDEA EN LA EDAD PEDIÁTRICA: A PROPÓSITO DE UN CASO. M. Doyle Sánchez, N. Ramírez Martínez, M. LL. González Castillo, M.J. Martínez García, L. García Villaescusa, R. Ruiz Cano.
Complejo Hospitalario Universitario de Albacete, Albacete.
Introducción: La orbitopatía tiroidea (OT) es un proceso autoinmune caracterizado por infiltración linfocitaria y edema de tejidos retrobulbares, secundario a inflamación del tejido graso y musculatura ocular extrínseca. Es infrecuente en la edad pediátrica, siendo rara por debajo de los 20 años, no existe predominio de sexo y la presentación clínica es menos grave que en adultos y púberes.
Motivo de consulta: Niña de 9 años presenta clínica compatible con hipertiroidismo, exoftalmos bilateral, bocio y temblor fino en manos, sin otra clínica acompañante.
Antecedentes familiares: Madre, 46 años hipotiroidea en tratamiento.
Exploración física: Peso 27, 8 Kg y Talla 135, 9 cm. FC 110 lpm. Bocio II con nódulo tiroideo en lóbulo izquierdo. Exoftalmos bilateral más llamativo en ojo derecho con movilidad ocular normal en todas las posiciones de la mirada. Prepuberal.
Pruebas complementarias: TSH:<0,01 µU/ml, T4L:7,77 ng/d,T3L :2, 78 ng/dl. TSI :29,3 mUI/ml.APO débilmente positivo 51, 75 UI/ml. Perfil lipídico: normal. ANA y EMA negativo. Ecografía tiroidea: Tiroides discretamente aumentado de tamaño con parénquima heterogéneo, de ecogenicidad aparentemente disminuida sin que se delimiten nódulos definidos. Gammagrafía tiroidea: bocio difuso hipercaptante sugestivo de enfermedad de Graves–Basedow. Edad ósea: 8 años y 6 meses a los 9 años y 1 mes.
Evolución: Al diagnóstico se inicia tratamiento con tiamazol
Comentarios: - En todo paciente con OT es prioritario estabilizar la función tiroidea y remitir al oftalmólogo para un seguimiento multidisciplinar.
- En aquellos casos de persistencia de OT a pesar de conseguir eutiroidismo, se recomienda tratamiento con corticoides. En primera instancia corticoides vía oral durante un mes y en caso de OT grave, glucocorticoides intravenosos a dosis altas como sucedió en nuestra paciente.
P1/d2-3-105 ¿ES EL METILFEDINATO CAUSA DE HIPERTIROTROPINEMIA? M. López García, C. Ontoria Betancort, S. Hernández Cáceres, A. Martínez Hernández, B. Reyes Millán, J.P. González Díaz.
Hospital de Motril (Granada).
Introducción: La hipertirotropinemia definida por aumento en plasma de TSH, siendo el resto de la función tiroidea normal, se ha relacionado con diversas causas: tiroideas (déficit de yodo, tiroiditis de Hashimoto, sobredosificación en el tratamiento de la enfermedad de Graves), no tiroideas (asociado a otras enfermedades, fármacos), así como con alteraciones genéticas (proteínas implicadas en la vía de la TSH).
Caso clínico: Varón de 9 años con antecedentes personales de vitíligo (tratamiento con Pimecrolimus tópico) y TDAH (Trastorno de Déficit de Atención e Hiperactividad) de reciente diagnóstico, en tratamiento con Metilfedinato oral (36 mg/día). Exploración física: máculas hipomelánicas en tronco y miembros inferiores, halo nevus en tórax anterior. Peso y talla en percentiles 75-90. Pruebas complementarias: Hematimetría y bioquímica normales, TSH 11,59 U/mL, T4F 1,31 ng/dL, Ac antitiroperoxidasa <10 UI/mL, Ac antitiroglobulina <20 UI/mL, Ac antiTSH 1,11 U/L, ecografía y gammagrafía tiroideas normales. Se asocia al tratamiento de base (Metilfedinato), L-tiroxina (50 mcgr/día), normalizándose los niveles de TSH (≤ 3,5 µU/L), tras múltiples controles. Tras 2,5 años de tratamiento, y ante la normalidad clínico-analítica se suspende la L-tiroxina, objetivándose tras esto, nuevo ascenso de tirotropina (14,61 U/L), con T3F, T4F y T4 total normales y anticuerpos negativos. Comentarios: Algunos estudios han propuesto una relación entre TDAH y disfunción tiroidea, especialmente resistencia a la hormona tiroidea. También se ha comunicado la relación del Metilfedinato con un leve descenso en la T4F y T4 total y leve aumento de TSH. En nuestro caso no podemos establecer con certeza absoluta la asociación entre TDAH-Metilfedinato y disfunción tiroidea (hipertirotropinemia). Pero al ser el resto de estudios de la función tiroidea normales, esto nos lleva a plantearnos esta posibilidad en el diagnóstico, aconsejándo el estudio de la función tiroidea antes del inicio y de forma evolutiva en los pacientes con TDAH en tratamiento con Metilfedinato.
P1/d2-3-106 HIPERTIROIDISMO NEONATAL: IMPORTANCIA DE LA COMUNICACIÓN ENTRE OBSTETRAS Y PEDIATRAS PARA SU DIAGNÓSTICO PRECOZ PERINATAL. A PROPÓSITO DE UNA OBSERVACIÓN. R. Briones Pascual, R. Pérez Iáñez, L. Olivares Sánchez, P. Cid Galache, J. Casas Gómez y JMª Gómez Vida.
Hospital de Motril, Motril (Granada).
Introducción: La patología tiroidea es relativamente frecuente en la gestante y es importante el reconocimiento precoz del trastorno en la embarazada para minimizar su posible repercusión en el desarrollo y crecimiento fetal. La enfermedad de Graves-Basedow (EGB) materna es la causa más frecuente de hipertiroidismo neonatal, estimándose que un 2% (0.6-9.6%) de sus recién nacidos desarrollarán hipertiroidismo.
Caso clínico: Recién nacida que a los 6 días de vida presenta: TSH <0.01 mUI/ml, T4L 6.67 ng/dl (1.8±0.3), T3 25.07 pg/ml (14.7±3.3), anticuerpos estimulantes del receptor de TSH 12.2 U/L (1.5-2.0), antitiroperoxidasa y antitiroglobulina negativos. Antecedentes: en historia ginecológica consta que la madre tiene un hipotiroidismo tratado hasta el 4º mes del embarazo con 125 microgramos de levotiroxina que se retira de forma paulatina por normalización de los niveles de hormonas tiroideas, quedando sin tratamiento desde el 5º mes. Realmente se trataba de una madre con EGB, tiroidectomizada subtotalmente 4 años antes, que mostraba una recaída de su enfermedad. Conocer este antecedente hubiera permitido un control más estrecho del feto y del recién nacido. Exploración: adecuado estado nutricional, llanto irritable, FC 200 - 240 lpm, temblores, tensión arterial: 97/54 mmHg, fontanela normotensa de 1cm x 1cm sin hiperostosis, sin más hallazgos patológicos. Se ingresa iniciándose tratamiento con propiltiouracilo (5,4 mg/kg/día) y propranolol (1.3mg/kg/día), con buena evolución clínica. Resto de pruebas complementarias, incluida ecografía tiroidea: normales. Evolución: a los 20 días se asocia levotiroxina (25mcg/día) por presentar: TSH 8.04 mUI/ml y T4L 0.53 ng/dl. Con 2.5 meses se negativizan los anticuerpos estimulantes del receptor de TSH. Se retira la medicación de forma completa a los 3 meses. La paciente presenta una ganancia ponderal en límite inferior de la normalidad con crecimiento craneal en límites normales y buen desarrollo psicomotor.
Conclusiones: Destacar la importancia del conocimiento de la causa de un hipotiroidismo materno al evaluar a su recién nacido y de la adecuada comunicación entre servicios (Obstetricia y Pediatría) para mantener un adecuado nivel de alerta en el diagnóstico de patologías como el hipertiroidismo neonatal, que no por infrecuentes tienen menor potencial de gravedad.
P1/d2-3-107 NEOPLASIA ENDOCRINA MÚLTIPLE TIPO 2B DE DIAGNÓSTICO TARDÍO. M.J. Olmos Jiménez( 1), M. Royo (1), B. Huidobro (1), C. Mata (1), E. Blarduni (2), M.D. Rodríguez-Arnao (1).
(1) Hospital General Universitario Gregorio Marañón, Madrid; (2) Hospital de Zumárraga.
Introducción: El carcinoma medular de tiroides es una enfermedad de alta mortalidad, que en la infancia suele aparecer en el contexto de los síndromes de neoplasia endocrina múltiple (MEN). En el MEN tipo 2B coexisten el carcinoma medular de tiroides junto con neuromas cutáneos en casi el 100% de los pacientes, el feocromocitoma en el 50% y hábito marfanoide. La penetrancia en el MEN2B llega casi al 100%, el 50% se trata de mutaciones de novo. Se recomienda realizar estudio de portadores y tiroidectomía profiláctica en los menores de 12 meses afectos. Sospechar esta enfermedad en niños con fenotipo compatible es de gran importancia debido su agresividad. Presentamos un caso de una niña diagnosticada de forma tardía en la que se inicia tratamiento con Sorafenib (inhibidor de Tirosin Kinasa) y su evolución.
Caso clínico: Niña de 10 años que ingresa para cirugía de epifisiolisis de cabeza femoral izquierda. Refería seguimiento en consultas de pediatría hospitalaria por retraso pondoestatural desde los 1,5 meses. No antecedentes familiares de interés.
Exploración física inicial: Peso de 22,2 Kg (-1,98DE) y talla 123,5 cm (-2.89DE), presentando labios prominentes y nódulos en mucosas (labio inferior, lengua y párpados). Resto de exploración normal incluida la zona cervical sin adenopatías ni masas palpables. Exploraciones complementarias: Calcitonina: 4475 pg/ml (0-14). Ecografía Tiroidea: Nódulos tiroideos con focos hiperecogénicos puntiformes. Aumento tamaño adenopatías cervicales. Proto-oncogen RET: Mutación Met918tre (exón 16)
Evolución: Se realiza tiroidectomía total y linfadenectomía cervical derecha y central (Mayo 2006). Anatomía patológica: Carcinoma Medular, afectación metastásica de adenopatías cervicales. Postcirugía: Octreoscan cervical positivo. Reintervención: linfadenectomía cervical bilateral, mediastínica y timectomía. Posteriormente continúa Calcitonina en aumento (hasta 13.000pg/ml). Enero 2010: PET-TAC con tenue actividad en región subcarinal y nódulos pulmonares milimétricos. Inicia tratamiento (uso compasivo) con Sorafenib, presentando como efecto secundario eritrodisestesia palmo-plantar. Tras 6 meses de tratamiento no progresión de la enfermedad y descenso de calcitonina hasta 2.620pg/ml.
Conclusiones: El tratamiento de elección del CMT es la tiroidectomía total profiláctica. En el MEN2B se recomienda realizarla antes de los 12 meses de edad por la baja supervivencia con los tratamientos habituales (QT, RT) en casos de diagnóstico tardío.
P1/d2-3-108 CÁNCER DE TIROIDES EN LA INFANCIA: PRESENTACIÓN DE TRES CASOS. C. Villalba Castaño(1), A. González Jimeno (1), A. Carcavilla Urquí (1), M. Güemes Hidalgo (1), M. A. Morlan López (2), A. Aragonés Gallego (1).
(1) Servicio de Pediatría. Hospital Virgen de la Salud, Toledo; (2) Servicio de Cirugía General. Hospital Virgen de la Salud, Toledo.
Introducción: El carcinoma diferenciado de tiroides es la neoplasia endocrina más frecuente en la infancia, aunque es una patología muy rara en este grupo (0.5-3% de los tumores pediátricos). El 72% son de tipo papilar. Entre 1990-2009 se declararon 76 casos de tumores tiroideos infantiles en España. Presentamos tres casos diagnosticados durante el año 2010.
Los principales datos se muestran en la tabla. Casos clínicos: Ningún paciente refería antecedentes de interés. En la ecografía tiroidea se objetivaron características de malignidad sugerentes de carcinoma papilar (nódulo sólido >1cm, heterogéneo con calcificaciones en su interior e intensa vascularización). El diagnóstico se confirmó con la PAAF y el estudio anatomopatológico. Para el tratamiento se realizó tiroidectomía total con vaciamiento cervical ipsilateral y del compartimento central. La paciente 3 presentó una mediastinitis al tercer día de la cirugía que precisó soporte ventilatorio y antibioterapia intravenosa de amplio espectro. Todos precisaron grandes aportes de calcio y vitamina D durante el postoperatorio.
Comentarios: El diagnóstico de tres casos de carcinoma papilar de tiroides en un año es inusual en nuestro medio y podría deberse al aumento de la incidencia que se describe en la literatura y cuyas causas no son bien conocidas. Como en nuestra serie, la ecografía de tiroides junto a la PAAF tienen gran rentabilidad en el diagnóstico. La alta prevalencia de hipoparatiroidismo postquirúrgico en nuestros casos podría deberse al abordaje quirúrgico actual más agresivo, así como a la edad de los pacientes. La presentación como enfermedad invasiva en los pacientes en que se ha realizado estudio de extensión se ha descrito con mayor frecuencia en la edad pediátrica.
P1/d2-3-109 CARCINOMA MEDULAR DE TIROIDES EN PACIENTE CON MEN 2B. L. Gutiérrez Pascual (1), A. De La Puente Arévalo (1), E. Bermúdez de Castro López (1), MA. Molina (1), I. González Casado (1), R. Gracia Bouthelier (1).
(1) Servicio de Endocrinología Infantil, Hospital Universitario La Paz, Madrid; (2) Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, IdiPAZ, UAM y CIBERER, ISCIII, Madrid.
El MEN 2B es una endocrinopatía producida por mutaciones activadoras del protooncogen RET (en el brazo largo del cromosoma 10), caracterizado por el desarrollo Carcinoma Medular de Tiroides (CMT) y feocromocitoma, sin hiperparatiroidismo. Es infrecuente (5-9% de los MEN 2) pero especialmente agresivo, y se manifiesta de forma precoz, asociando un fenotipo característico. Presentamos un varón de 14 años con los siguientes antecedentes personales: RNT de peso elevado para su EG que presentaba pequeñas lesiones mucosas en lengua desde el período neonatal; durante la infancia aparecen lesiones similares en labios, que relacionan con traumatismos, dado que presentaba pies cavos y tendencia a la hipotonía con caídas frecuentes. A partir de los 6 años observan cambios faciales, con mayor prognatismo e hipertelorismo, y a partir de los 11 años escoliosis y aumento de talla llamativo sin ganancia de peso, coincidiendo con el inicio puberal. En ese momento comienzan a notar pequeños “bultos” cervicales dolorosos, con aumento progresivo de tamaño, que fueron diagnosticados de adenopatías inflamatorias. Finalmente a la edad de 14 años, ante un fenotipo compatible con síndrome MEN 2B, se realiza estudio genético, hallándose la mutación M918T en heterocigosis en el exón 16 del gen RET, que confirma el diagnóstico. Tras estudio bioquímico y de imagen compatible con CMT se realiza tiroidectomía total con vaciamiento cervical, siendo imposible la resección tumoral total por extensión a estructuras vecinas.
En la mayoría de los MEN 2B la mutación se presenta de novo, siendo así en nuestro paciente, por lo que el fenotipo constituye la única pista para poder hacer un diagnóstico precoz. En función del tipo de mutación estaría indicado realizar tiroidectomía profiláctica con mayor o menor precocidad (en la aquí hallada antes incluso del año de edad, debido a su agresividad), pero en cualquier caso la resección total supone el único tratamiento curativo del CMT, que aparecerá en el 100% de los casos.
Debemos sospechar, pues, un MEN 2B en todo paciente con ganglioneuromas orolabiales, conjuntivales o intestinales, como rasgo característico, especialmente si se asocian con hábito marfanoide, aunque no presenten antecedentes familiares y realizar estudio genético para confirmarlo.
P1/d2-3-110 HEMIAGENESIA TIROIDEA COMO CAUSA INFRECUENTE DE HIPOTIROIDISMO SUBCLÍNICO. L. Salamanca Fresno, M. A. Molina Rodríguez, A. de la Puente Arévalo, A. C. Barreda Bonis, I. González Casado, R. Gracia Bouthelier.
Servicio de Endocrinología Pediátrica. Hospital Universitario La Paz. Madrid.
Introducción: La hemiagenesia tiroidea es un anomalía congénita de la glándula tiroidea consistente en la ausencia o hipoplasia severa de un lóbulo (volumen < 1/10). La verdadera prevalencia se desconoce dado que la mayoría de las veces transcurre de forma asintomática, aunque se estima en torno a 0,05 – 0,2%. Material y Métodos: Presentamos 3 casos clínicos de pacientes pediátricos con diagnóstico de confirmación de hemiagenesia tiroidea por medio de gammagrafía con trazador 99mTc.
Resultados: Los 3 pacientes se mantenían clínicamente asintomáticos. El motivo del diagnóstico en el primer paciente fue una elevación transitoria de la TSH con motivo de estudio por retraso pondero-estatural. En el segundo paciente una ecografía cervical por estudio de adenopatías inflamatorias constata una posible ausencia de lóbulo tiroideo. El tercer paciente fue diagnosticado en el cribado neonatal. En los 3 casos el diagnóstico se confirmó por medio de gammagrafía con 99mTc. Dos pacientes carecían del lóbulo izquierdo y el paciente restante carecía del derecho. Los pacientes 1 y 3 han recibido tratamiento sustitutivo, el paciente 1 de forma transitoria y el 3 de forma permanente. Discusión: La etiología de la hemiagenesia tiroidea todavía se desconoce. Confirmando nuestros resultados la mayoría de los estudios la refieren como más frecuente en el lóbulo izquierdo y en mujeres. Aunque se describe en la literatura como una condición benigna, la mayoría de los pacientes afectos asociaban alteraciones tiroideas en la edad adulta siendo el más frecuente el bocio nodular. Aunque suele confirmarse por medio de gammagrafía, la ecografía sigue siendo una buena herramienta diagnóstica por su reproductibilidad y coste-efectividad.
P1/d2-3-111 PSEUDOHIPOPARATIROIDISMO EN MADRE E HIJO. V. Cancela Muñiz(1), E. Artola Aizalde (1), E. Blarduni Cardon (2), O. Sardon Prado (1).
(1) Hospital Donostia. San Sebastián, (2) Hospital de Zumárraga. Zumárraga.
Caso clínico. Motivo de Consulta: Paciente remitido a consulta de Endocrinología Infantil a los dos meses desde la Unidad Neonatal por hipotiroidismo.
Antecedentes obstétricos y familiares: Embarazo normal. Parto por cesárea en semana 37+5 de gestación. PN: 2.820g. L: 44 cm. Madre 30 años, fenotipo Albright. Obesidad mórbida intervenida. Estudiada en la infancia por baja talla sin diagnóstico etiológico. Padre sano.
Antecedentes personales: Ingreso al nacimiento por distrés respiratorio. Los primeros días precisa tratamiento con calcio por hipocalcemia (7,1 mg/dl), normalización posterior. Analítica a los 13 días de vida: hipotiroidismo con T4L 0,88 ng/dL y TSH 24,02 mUI/L con inicio de tratamiento con L- Tiroxina (screening neonatal de hipotiroidismo normal).
Evolución: El paciente es controlado en la consulta por hipotiroidismo presentando buen control con tratamiento con L- Tiroxina. Los niveles de calcio se mantienen dentro de la normalidad. Exploración: cara redonda, talla corta y obesidad. Debido al fenotipo materno y del niño, se realiza estudio de biología molecular estudiándose en gen GNAS del cromosoma 21q13, con resultado de mutación a nivel del axon 1, consistente en c.8G>A; p.Cys3Tyr, compatible con pseudohipoparatiroidismo en el niño. La madre presenta la misma mutación.
Comentarios: El paciente presenta un pseudohipoparatiroidismo, con fenotipo Albright y resistencia a PTH y TSH (con hipotiroidismo en tratamiento con L- Tiroxina), con calcio normal actualmente y probable afectación futura de este. Esta forma se da cuando la mutación se hereda de la madre. La madre presenta un pseudopseudohipoparatiroidismo con fenotipo compatible sin afectación hormonal.
P1/d2-3-112 CARCINOMA DE TIROIDES EN EDAD PEDIÁTRICA: NUESTRA EXPERIENCIA. M.L. Bertholt, C. Luzuriaga Tomas, C. Freijo Martín. J.L. Guerra Díez, D. Casanova Rituerto.
Unidad Endocrinología Pediátrica. Unidad de Cirugia general. Hospital Marqués de Valdecilla, Cantabria.
Introducción: El carcinoma de tiroides en edad pediátrica, es poco frecuente. Predominio en mujeres, importante por su gravedad. Representa el 1,4% de las neoplasias infantiles. La variedad histológica dominante es la papilar.
Material y Métodos: Analizamos las historias de 2 pacientes con carcinoma papilar de tiroides, diagnosticados durante los 35 años de experiencia de nuestro servicio. Resultados: Caso1: controlada desde 5.6 años por pubarquia precoz aislada, obesidad, resistencia insulina. Con 10.3 años se detecta ”nódulo tiroideo”. T4libre (1,40 ng/dl), TSH (0,28 mUI/ml), Tirogolbulina(1.110 ng/ml). Ecografía: lóbulo derecho tumoración 2,5x1,5x1,5; hipoecoica, rodeada de halo hipoecoico, zonas quísticas en su interior. Gammagrafía Tc99m/ I-131: nódulo hipercaptante en lóbulo tiroideo derecho. Cirugía (10,8 años).Biopsia intra-operatoria: lóbulo tiroideo derecho carcinoma papilar patrón folicular, infiltración focal de la cápsula; lóbulo tiroideo izquierdo carcinoma papilar microscópico no encapsulado. Completada cirugía total sin observarse adenopatías. Evolución 9,5 años, rastreó corporal con TL201/I-131 tras 4 semanas, estimulando TSH endógena, focos de captación en tiroides con Tiroglobulina (5,4 ng/ml), se administra 30 mCiv.o de I-131, rastreos periódicos, con 6 meses persiste mínimo tejido, repiten dosis de 30 mCiv.o de I-131, posteriormente anual, tras 4 años se aprecia captación en tejido tiroideo repitiendo dosis de 30 mCiv.ode I-131. No más problemas.
Caso 2: Controlada desde 5.3 años por hipotiroidismo subclínico, tratada con levotiroxina a 1,3 mcg/día. Madre: nódulos tiroideos intervenidos (AP: hiperplasia nodular). Ac-antitiroideos negativos, ecografías tiroideas normales, pubertad adelantada, menarquia 10.75 años. Tras retirar levotiroxina, 6 meses, TSH alta, palpación: bocio, predominio lóbulo tiroideo derecho y nódulo de consistencia dura. Tiroglobulina(1.226 mcg/ml),T4L(0,97 mcg/dl),(TSH 3,55 mUI/ml). Ecografía tiroidea: múltiples nódulos distribuidos por ambos lóbulos. Gammagrafía: nódulo hipocaptante dominante en seno de bocio multinodular con nódulos de variada actividad funcional. Tiroidectomía total (12,3 años), ganglios normales. AP: carcinoma papilar bilateral, multifocal, predominio de patrón folicular, no infiltra tejido circundante. A las 4-6 semanas rastreo corporal I-131/MIBI marcada con Tc99m: restos tiroideos en lecho tiroideo, niveles de Tiroglobulina indetectables; se administra 30 mCiv.o de I-131, evolución 6 meses normal.
Conclusiones: El cáncer diferenciado de tiroides, patología infrecuente pero importante, debe ser diagnosticado precozmente, con un examen correcto del cuello. Requiere protocolizar la actuación diagnóstica, de tratamiento y seguimiento. El tratamiento inicial será agresivo y extenso, porque condiciona la evolución, evitando diseminación linfática y pulmonar.
P1/D2-3-113 TEST DE TRH EN LA DISCRIMINACIÓN ETIOLÓGICA DEL HIPOTIROIDISMO CENTRAL. J.C. Moreno(1), A. De la Puente (2), J. Guerrero (2), R. Mora (3), J.M. Iturzaeta (3), R. Gómez (3), A. Buño (3), M.A. Molina (2), R. Gracia (2).
(1) Laboratorio de Tiroides Molecular. INGEMM Instituto de Genética Médica y Molecular. Hospital Universitario La Paz. Madrid; (2) Servicio de Endocrinología Pediátrica. Hospital Universitario La Paz. Madrid; (3) Servicio de Bioquímica y Análisis Clínicos. Hospital Universitario La Paz. Madrid.
El Hipotiroidismo Congénito Central (HCC) tiene una prevalencia de 1:16.000 nacidos. En un 40% es una deficiencia aislada, ya hipofisaria, ya de origen hipotalámico. El HCC queda fuera del screening neonatal de HC, pues mayoritariamente se utilizan los niveles de TSH como método de despistaje. Tras un diagnóstico tardío, no es rutina el discriminar entre el HCC secundario y terciario con el test de TRH. El HCC hipofisario se debe a mutaciones de los genes TSHB y TRHR, mientras que el hipotalámico es genéticamente “huérfano” en humanos.
Objetivo: Testar la capacidad discriminativa del test de TRH en la etiopatogénia del HCC.
Métodos y Pacientes: Tras 7 µg/kg de TRH determinamos TSH y PRL a los -15, 0, 15, 30, 45, 60, 120 y 180 min, y T4L y T3T a los 0 y 180 min. Se analizaron tanto el pico neto de TSH y su retorno completo a valores basales como la dinámica de su incremento (ratios 15´/0´ y 30´/0´) y descenso (ratios 30´/60´ y 180´/0´) según Van Tijn.
El test se realizó en: 1. Niña de 20 meses con retraso pondero-estatural postnatal progresivo severo (-3,7 SD peso y talla) con hiperfagia y TSH reducida respecto de los niveles de T4 (TSH 1,2 mU/L, T4L 0,92 ng/dl). 2. Niño de 12 años con crisis de hipotermia-bradicardia-sudoración e hipertirotropinemias esporádicas (TSH 5,4-8 mU/L) y 3. Niño de 11 años con hipertirotropinemia (TSH 9,82 mU/L) e hiperglucemia (109 mg/dl) (sospecha defecto TRH).
Resultados: Los pacientes 1 y 2 tienen una respuesta tipo 3 (defecto hipotalámico) con ratios 15´/0´ de 23,8 y 9,1 (N<6.5) respectivamente, sin retorno a la basal tras 3 horas (ratios 180´/0´ de 2.87 y 1.7). El paciente 3 tuvo una respuesta tipo 0 (hipotiroidismo primario) con ratio 15´/0´ de 4.6 y 180´/0´ de 0.8.
Conclusiones: El test de TRH de 180 min. identifica defectos hipotalámicos del eje tiroideo incluso en pacientes con eutiroidismo bioquímico. Los hipotiroidismos terciarios pueden cursar tanto con elevación como con disminución relativa de TSH (que puede estar en rangos normales), lo que sugiere la utilidad del “índice de TSH” en las consultas de endocrinología. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites