| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):221-229 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.191 |
| Tiroides |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
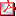 Tabla - P2/d3-119 Tabla - P2/d3-119 |
| Tabla - P2/d3-125 |
| Tabla - P2/d3-127 |
| Figura - P2/d3-129 |
|
|
P2/d3-119 EVALUACIÓN DE LA HIPERTIROTROPINEMIA EN UNA CONSULTA DE ENDOCRINOLOGÍA INFANTIL M.J. Alcázar Villar, M.J. Rivero Martín, M. Sanz Fernández, C. Navarro Moreno Hospital Universitario de Fuenlabrada. Servicio de Pediatría, Fuenlabrada. Madrid Introducción: La elevación de la TSH en los niños no siempre refleja la existencia de hipofunción tiroidea. En muchas ocasiones es un hecho aislado en sujetos asintomáticos o con síntomas inespecíficos, por lo que actualmente parece más adecuado el nombre de hipertirotropinemia. Objetivo/métodos: Determinar el tiempo de normalización de los valores de TSH, la evolución a hipotiroidismo, causa de solicitud de analítica y síntomas y tratamiento instaurado. Para ello llevamos a cabo un estudio retrospectivo de los pacientes entre 1 mes y 15 años derivados por elevación de TSH (5,5-10 μU/ ml), atendidos en consulta desde enero de 2010 a diciembre de 2012. Analizamos: edad, sexo, índice de masa corporal (IMC), síntomas que motivaron la consulta, valores de TSH y T4L al diagnóstico y en controles posteriores. Resultados: Se analizan datos de 80 pacientes. La relación varón: mujer fue 1:1. La edad media 7,5 años (rango 1-13.8a), siendo el 7,5% menores de 2 años. Cifra media de TSH al diagnóstico: 7μU/ml (DE: 0,88) con T4L media: 1,21ng/dl (DE: 0,05). El motivo de solicitud del perfil tiroideo estuvo relacionado con el peso (bajo peso o sobrepeso) en el 30% de los casos, siendo la sintomatología muy dispar en el resto (Tabla 1). Al diagnóstico no se instauró tratamiento en ningún paciente. Se realizó analítica control entre 15 días y 11 meses (media 4,6 meses). En el 68,75% de los pacientes se normalizó la cifra de TSH en el primer control mientras que en el 31,25% persistía TSH elevada con T4L normal. De este 31,25% normalizaron valores posteriormente el 47%, se inició tratamiento con levotiroxina en 2 y el resto mantienen controles periódicos de TSH-T4L. Por tanto sólo el 13,75% de todos los pacientes estudiados continúan seguimiento. Comentarios: La hipertirotropinemia es frecuente en la población infantil, afectando en nuestro caso por igual a ambos sexos. La determinación rutinaria de hormonas tiroideas en el estudio de patologías no relacionadas con el hipotiroidismo puede poner de manifiesto una elevación de TSH con T4L normal no relacionada con patología tiroidea. Importancia de seguimiento de pacientes con hipertirotropinemia para constatar normalización o la evolución a hipotiroidismo subclínico.
P2/d3-120
DISFAGIA, SIN BOCIO COMPRESIVO, COMO FORMA DE PRESENTACIÓN DE HIPOTIROIDISMO M. López Capape(1), M.J. Cusco Fernández(1), M.J. Tellez Kling(2), J. Lujan Martinez(3), I. Alonso Martinez(3), J. Jimenez Martinez(3) (1) U. Endocrinología Pediátrica. Servicio de Pediatría Hospital La Moraleja, Sanchinarro. Madrid; (2) Centro Medico Milenium Alcobendas; (3) Servicio de Pediatría H. La Moraleja Introduccion: La tiroiditis linfocitaria crónica constituye la patologia tiroidea mas frecuente en el niño y adolescente. Puede cursar con hipotiroidismo, y en estos casos, las manifestaciones mas habituales son bocio y retraso en el crecimiento. Excepcionalmente hay dolor local o disfagia por mecanismo compresivo del tiroides aumentado de tamaño, pero se han descrito alteraciones en la peristalsis esofágica en pacientes con hipotirodismo grave, incluso en ausencia de bocio. Presentamos un caso de hipotiroidismo con disfagia como unica expresividad clinica. Caso clinico: Niña de 9 años sin antecedentes de interés, que presenta disfagia de un mes de evolucion. Refieren leve pérdida de peso, no cuantificada, por negarse a comer sólidos. No refieren desaceleración en crecimiento, cambios en ritmo intestinal, intolerancia al frío ni astenia. No nota molestias cervicales ni aumento de tamaño del cuello. Valorada por ORL con faringoscópia y esofagograma normales se remite para estudio. EF: Talla 145,5 cm (p89), TD: 170 cm (p84). Peso 33,6Kg. (p42). IMC 15,8 Kg/m2 (p21). TA 113/66. Fc 64lpm. BEG, piel seca, bocio grado I. ACP normal. Abdomen normal. GU Tanner 1. P. Complementarias: Se realiza analitica en la que destaca hipercolesterolemia de 258mg/dl y alteracion de la funcion tiroidea: TSH 108,5 (0,7-5,7mU/L) , T4 6,1 (5,2-10ug/dl), T4L 0,8 (0,8-1,8ng/dl), T3 1 (0,8-2,1ug/L). Confirmandose el hipotiroidismo: TSH 89,9mU/l, T4L 0,8 ng/dl, T3L 2,62 (3-6ng/L ), con Ac anti-TG positivos (103,8UI/ml, CN<60). En la ecografia tiroidea se describe un tiroides de morfologia normal, LD: 16x14x28mm y LI 13x11x32mm, de contornos discretamente lobulados y ecogenicidad algo disminuida y marcado aumento de vascularización. Evolución: se inicia tratamiento con levotiroxina a dosis habituales, apreciando una clara mejoria clinica y analitica en controles posteriores. Conclusiones: La disfagia por alteracion de la peristalsis esofágica, puede aparecer como sintoma aislado en niños con hipotiroidismo, aun en ausencia de bocio.
P2/d3-121
ENFERMEDAD DE GRAVES BASEDOW DE INICIO TEMPRANO EN PACIENTE CON SÍNDROME DE WISKOTT ALDRICH M.V. Bovo, G. Bueno Lozano, A. Ayerza Casas, F. Ramos Fuentes, J.L. Olivares López, J.M. Garagorri Otero Servicio de Pediatría. Hospital Clínico Universitario Lozano Blesa. Zaragoza, Zaragoza Introducción: El síndrome de Wiskott-Aldrich (WAS, OMIM 301000) es una enfermedad rara con una incidencia 1:100.000 nacidos vivos. Se trata de una inmunodeficiencia primaria con herencia ligada al cromosoma X, debida a mutaciones en el gen que codifica la proteína (WASP) con manifestaciones clínicas diversas. La incidencia de autoinmunidad en los pacientes con WAS es alta (40-72%), aunque su causa no está bien definida. Citas bibliográficas recientes han comunicado que los trastornos autoinmunes en edad pediátrica más frecuentemente asociados a WAS son: anemia hemolítica, vasculitis, artritis, neutropenia, enfermedad inflamatoria intestinal, glomerulonefritis por IgA, entre otros. Caso clínico: Paciente diagnosticado de síndrome de Wiskott-Aldrich a los 17 meses de edad, (mutación missense (R86H) en el exon 2 del cromosoma Xp11). A los 20 meses recibe tratamiento curativo mediante transplante de médula ósea alogénico (procedente de hermano HLA-idéntico) con evolución clínica satisfactoria. A los 3 años y 3 meses inicia clínica sugerente de hipertiroidismo (exoftalmos bilateral, bocio, taquicardia e insomnio), confirmándose diagnóstico de Enfermedad de Graves-Basedow (TSH: < 0,003 microU/mL (0,280-2,640 microU/mL), T3L: 28,2 pg/ mL (1,9–3,4 pg/mL), T4L: 5,28 ng/dL (0,9–1,7 ng/ dL), Anticuerpos antitiroides microsomal: 773 UI/ mL (0–28 U/mL), Anticuerpos antitiroglobulina: 994 UI/ml (0-70UI/mL), Anticuerpos antireceptor TSH (TBII): 335,7% (<0,7%). Ecografía tiroidea: Bocio difuso. Gammagrafia tiroidea: Tiroides aumentado de tamaño con distribución homogénea del trazador). Se inicia tratamiento con Metimazol (dosis: 0,5 mg/Kg/día) aumentandola progresivamente hasta la normalidad clínica y analítica; y posteriormente se disminuye hasta suspenderla por hipofunción tiroidea. Desde los 12 años de edad hasta la actualidad (17 años) permanece en normofunción tiroidea bajo tratamiento con Levotiroxina (150 microgr/día). Los anticuerpos anti-tiroideos persisten muy elevados (Anticuerpos antitiroides microsomal: > 3.000 UI/mL, Anticuerpos antitiroglobulina: > 9.000 UI/ml). Comentarios: En este caso llama la atención el inicio de la Enfermedad de Graves-Basedow a una edad temprana (3 años, 3 meses), y 20 meses después del trasplante de médula ósea, tratamiento que, para algunos autores, debería de haber sido curativo del trastorno inmunológico y evitado el desarrollo de enfermedad autoinmune.
P2/d3-122
DISHORMONOGÉNESIS TIROIDEA DE GENÉTICA NO FILIADA EN FAMILIA CON TRES HERMANOS AFECTOS E. Maqueda Castellote(1), J.C. Moreno Navarro(2), A. Iglesias Alvarez(3), J. Sáchez Pérez(4), D. Belver Garcia(4), R. Coripio Collado(4) (1) Servicio de Pediatria. Hospital Sabadell.Corporació Sanitaria Parc Taulí. Institut Universitari Parc Taulí-UAB, Sabadell. Bacelona; (2) Laboratorio Molecular de Tiroides. Hospital Universitario La Paz. Madrid; (3) Laboratorio Molecular de Tiroides. Instituto de Genética Médica y Molecular(INGEMM) del Hosital Universitario La Paz. Madrid; (4) Hospital Sabadell.Corporació Sanitaria Parc Taulí. Institut Universitari Parc Taulí-UAB. Sabadell. Barcelona Introducción: La dishormonogénesis tiroidea familiar es un tipo de hipotiroidismo congénito (HC) primario que consiste en un bloqueo total o parcial de los procesos bioquímicos implicados en la síntesis y secreción de hormonas tiroideas. Constituye el 10-20% de la etiología global del HC. Suele presentar un patrón autosómico recesivo. Presentamos un caso de dishormonogénesis en una familia consaguínea. Caso clínico: Recién nacido varón de 7 días remitido por sospecha de hipotiroidismo congénito. Antecedentes familiares: Padres sanos marroquíes y consanguíneos (primos hermanos), hermanos de 17 y 11 años diagnosticados a los 8 meses y 2 meses de edad respectivamente de HC en Marruecos. El mayor presenta retraso mental. Ambos presentan nódulos <1cm en la ecografía de tiroides. No antecedentes de bocio. Familia consumidora de sal marina. Evolución: A las 48 horas de vida se cursa analítica sérica coincidiendo con el cribaje neonatal que muestra TSH 881, T4 libre de 0,15 (1,8-4,1). Se inicia tratamiento con levotiroxina a 30 mcg/día. Previo al inicio de tratamiento se extrae muestra para ampliar estudio: Tiroglobulina>300ng/mL( 0,83-68), anticuerpos antiperoxidasa tiroidea y tiroglobulina negativos. La ecografía de tiroides muestra tiroides ortotópico y algo aumentado de tamaño. Radiografía de rodilla normal. La gammagrafía con I123 y el test de perclorato es positivo (descenso de captación del 12,61%) indicando alteración de la organificación de yodo. Paciente asintomático. Se ajusta tratamiento según niveles de TSH y T4 libre. Yoduria baja en todos los familiares (72-178 mcg/g creatinina (N: 825-2950 mcg/g creatinina). Estudio genético: Cursado ante la sospecha de dishormonogénesis tiroidea familiar en los genes DUOX2, DUOXA2 y DEHAL1 a la familia. En DEHAL1 se identificó un cambio en heterozigosis en el hermano mayor, menor y el padre (p.Arg246Gln =c.737A/G) que 2/4 programas de predicción de patogenicidad clasifican de posiblemente dañino. En el estudio de DUOX2 se identificaron varios SNP (p.H678R, p.R701Q y p.S.1067L) que no cosegregan con el fenotipo hipotiroideo. Conclusiones: El estudio genético tiroideo, en ausencia de defectos bialélicos en los pacientes sugiere el estudio de otros genes que participen en la biosíntesis tiroidea o de técnicas más complejas (Array-CGH) para completar la filiación genética a este caso.
P2/d3-123
ALOPECIA AREATA COMO POSIBLE MANIFESTACIÓN NO ENDOCRINA DE LA ENFERMEDAD DE GRAVES-BASEDOW P. Romero García, C. Villalba Castaño, M.J. González Iglesias, E. Arrontes Caballero Hospital San Rafael, Madrid Introducción: La enfermedad de Graves-Basedow (EGB) es la causa más frecuente de hipertiroidismo en la población pediátrica aunque su prevalencia es baja 0.02%. Se caracteriza por clínica de hipertiroidismo, bocio difuso y oftalmopatía. Puede asociarse a otras enfermedades de etiología autoinmune, como es en nuestro caso clínico la alopecia areata (AA). La AA es una enfermedad del cabello no cicatricial de etiología autoinmune. Caso clínico: Niña de 6 años y 11 meses que acude por clínica compatible con hipertiroidismo (pérdida de peso, hipersudoración, hiperdefecación, hiperactividad, mal rendimiento escolar, problemas de atención) de 2 meses de evolución asociado a exoftalmos bilateral y bocio. AF. Padre: DM tipo I. Rama materna y hermano: hipotiroidismo autoinmune. AP: Dermatitis atópica. EF: Peso: 23.5 kg(p41). Talla: 129.8 cm(p93). IMC: 13%(p14). TA: 91/70 mmHg. Fc 123 lpm. Bocio II. Exoftalmos bilateral con movilidad ocular normal en todas las posiciones de la mirada. Tanner I. Pruebas complementarias: Laboratorio: TSH <0,03 uUI/ml, T4libre: 4,25 ng/dl. T3libre: 23.6 ng/ dl. Ac anti-tiroideos: anti TG: 765 UI/ml; anti TPO 724 UI/ml; anti TSI 4,38 UI/l. Ecografía tiroidea: tiroides de tamaño difusamente aumentado sin identificar nódulos predominantes. Hipervascularización. Ecocardiografía: sin alteraciones. Edad ósea: 8 años. Evolución: Se inicia tratamiento con propanolol a 1,5 mg/kg/día las 3 primeras semanas y metimazol a 0,5 mg/kg/día. Al 1,5 meses se añade levotiroxina. Normalización de la función tiroidea y negativización de los Ac anti TSI a los 3 meses. A los 3 meses presenta placas de alopecia redondeadas, asintomáticas, de aparición brusca en la región parieto-occipital bilateral compatibles AA. Actualmente, a los 6 meses de evolución, persiste bocio Ib y exoftalmos bilateral. En tratamiento con metimazol a 0,15 mg/kg/día. Las placas de AA han ido en aumento con mala respuesta al tratamiento convencional. Discusión: La AA es una patología infrecuente en la edad escolar, siendo su incidencia asociada a la EGB muy rara, no encontrando apenas casos descritos en la literatura. Está asociada a otras enfermedades autoinmunes. Son factores de mal pronóstico el inicio a edad prepuberal, la asociación a alteraciones tiroideas, la forma clínica ofiásica así como su presencia en pacientes con dermatitis atópica.
P2/d3-124
OVARIOS MULTIQUÍSTICOS EN NIÑA CON HIPOTIROIDISMO PRIMARIO V. Cancela Muñiz, E. Artola Aizalde, N. Pacho Beristain Hospital Universitario Donostia/Pediatría, Gipuzkoa Motivo de consulta: Paciente mujer de 12 años que consulta en Urgencias por dolor abdominal continuo de 2 días de evolución. Cefaleas intensas desde hace dos semanas. Ausencia de reglas en los dos meses previos. Antecedentes personales: Valorada a los 10 años en Consulta de Neurología Pediátrica por cefaleas, dada de alta con TC craneal normal. Menarquia con 10 años. Antecedentes familiares: Madre y tía hipotiroidismo primario. Exploración física: Peso: 60 Kg (P>97). Talla: 150 cm (P50-75). IMC: 26,6%. Distensión abdominal. Dolor en hemiabdomen derecho a la palpación. Resto normal. Pruebas complementarias: Ecografía doppler abdominal: ocupando abdomen tumoraciones quísticas multilobuladas, bien definidas, pared y tabiques regulares. Tamaño 15 -18 cm cada una, muy probable origen ovárico. Sugieren carácter benigno. Analítica: Hemograma, bioquímica, coagulación normal. Metabolismo del hierro y transaminasas normal. Ca125: 145,7 U/ml (0-35). Alfafetoproteína y Ca 19,9 normal. RM abdominal: gran masa abdominal bilobulada de 20X10X21 cm, con tabiques finos en su interior y pequeñas áreas focales con líquido. Epicentro en abdomen sin poder descartar origen ovárico o peritoneal. Se realiza una RM cerebral para descartar tumor cerebral con resultado de hiperplasia hipofisaria (15,5 mm de diámetro mayor cráneo caudal). Resto sin alteraciones. Se completa analítica: T4L 0,10 ng/dl, TSH > 500 mU/L. FSH: 7,4 U/L, LH: 0,1 U/L, Estradiol: 1.307 pg/ml, prolactina 109,4 ng/ml. Ca 125: 137 U/ml. Diagnóstico: Tras el resultado analítico se diagnostica de ovarios multiquísticos debido a hiperestimulación por hipotiroidismo primario. Tratamiento: Inicia tratamiento con Levotiroxina: 25 mcg/día y gestodeno+etinilestradiol. Evolución: Normalización de la función tiroidea, de niveles de LH y estradiol , del tamaño ovárico y uterino y del tamaño hipófisiario. Conclusiones: Existe una similitud entre FSH, TSH y sus receptores. Grandes cantidades de TSH pueden estimular el R de FSH. Además existe mayor sensibilidad a FSH de los ovarios pre o peripuberales. Debido al aumento de TSH se produce una hiperplasia hipofisaria que puede confundirse con patología tumoral. Ante la presencia de ovarios con múltiples quistes es importante una valoración endocrinológica para evitar posibles tratamientos incorrectos.
P2/d3-125
HIPERTIROIDISMO EN PACIENTES PREPUBERALES ¿QUÉ SABEMOS? A. González Vergaz(1), B. García Cuartero(1), J.L. Ruibal Francisco(2), R. Ruiz Cano(3), J. Ramirez Fernández(4), M.J. Ceñal González- Fierro(5), P. Gutierrez Díez(6) (1) Hospital Universitario Severo Ochoa. Endocrinología pediátrica., Leganés/Madrid; (2) Hospital Universitario Infanta Cristina. Parla; (3) Hospital General Universitario. Albacete; (4) Hospital Universitario Príncipe de Asturias. Alcalá de Henares; (5) Hospital Universitario Móstoles.Móstoles; (6) Hospital Universitario de Getafe. Getafe Introducción: El hipertiroidismo es infrecuente en la edad pediátrica,la sintomatología es insidiosa, frecuentemente neuropsiquiátrica, con peor pronóstico a edades tempranas. Objetivo: Describir las características clínicas, bioquímicas y la respuesta al tratamiento en una población de pacientes prepuberales con hipertiroidismo. Material y métodos: Estudio retrospectivo, descriptivo, multicéntrico de pacientes diagnosticados entre 1993-2007. Se recogieron datos clínicos y bioquímicos al diagnóstico y durante la evolución. Los resultados se analizaron con SPSS.15 Resultados: Veintidos pacientes, 16 niñas (72,7 %) , edad media 8,79 (2,08) años , entre 4 y 12,1 años, tres menores de cinco años, todas ellas niñas. Inician síntomas con 8,39 (2,29) años. Las características clínicas y bioquímicas están reflejadas en la Tabla 1. Presentaron reacciones adversas leves el 22,7 % (40 % menores de 5 años). Catorce recaídas en nueve pacientes, que continuan tratamiento hasta 5,58 (3,2) años . Nueve pacientes (77,7 % niñas) remiten, pero sólo una de las que prolongó tratamiento; cinco precisan tratamiento quirúrgico, dos tratamiento con Yodo, cuatro continuan metimazol y dos abandonan. Conclusiones: -El diagnóstico se realizó precozmente. -La distribución por sexos, la sintomatología y la positividad de los anticuerpos es similar a lo descrito en puberales. -La edad ósea es superior a la cronológica. -Las reacciones adversas son más frecuentes a menor edad. -La remisión con tratamientos prolongados es similar a otras edades, pero si hay recaídas, mantenerlo no mejora el pronóstico y deberíamos plantear otros tratamientos.
P2/d3-126
HIPOTIROIDISMO ADQUIRIDO EN EL PERÍODO NEONATAL M. González Fernández-Palacios, M.D. Palomar, A.C. García-Martínez, T. Gallegos, F. Camacho, E. García-García Hospital Virgen del Rocio / Endocrinología Pediátrica, Sevilla Introducción: No todos los hipotiroidismos que se presentan en el periodo neonatal son de origen congénito. Hay algunas circunstancias que producen déficit adquirido de la función tiroidea y, por tanto, no son detectados por el despistaje universal. Los hemangiomas son tumores benignos que producen hipotiroidismo de consumo debido al incremento de la actividad de la enzima iodotironina deidodinasa tipo 3. Esta enzima cataliza la conversión de tiroxina (T4) a triyodotironina inversa (T3r), sustancia inactiva, e inhibe la producción de triyodotironina (T3), la hormona activa. Caso clínico: Lactante de 1 mes remitido desde oncología por valores elevados de hormona tirotropa (TSH) (86 mcU/ml) tras estudio por hemangiomatosis hepática y cutánea. Inicia tratamiento sustitutivo con levotiroxina y para los hemangiomas recibe propranolol. A los dos meses presenta una disminución del tamaño de los hemangiomas a la vez que se puede bajar la dosis de levotiroxina hasta suspender a los cinco meses de tratamiento. Conclusión: El hipotiroidismo de consumo es una manifestación paraneoplásica de los hemangiomas que debemos diagnosticar y tratar precozmente para preservar el desarrollo neurológico. El paralelismo entre la extensión tumoral y la actividad inhibitoria sobre las hormonas tiroideas permite ir retirando el tratamiento sustitutivo cuando se constata regresión tumoral.
P2/d3-127
HIPERTIROIDISMO NEONATAL EN HIJOS DE MADRE CON ENFERMEDAD DE GRAVES TRATADAS CON RADIOYODO A.C. García Martínez, F. Camacho, T. Gallegos, M. González Fernández-Palacios, M.D. Palomar, E. García-García Hospital Virgen del Rocio / Endocrinología Pediátrica, Sevilla Introducción: Fetos y neonatos de madres con enfermedad de Graves pueden sufrir hiper o hipotiroidismo según el balance entre anticuerpos estimuladores e inhibidores frente al receptor de la tirotropina (Ac RTSH) y fármacos antitiroideos que reciben a través de la placenta. Aunque el hipertiroidismo es autolimitado, puede comprometer la vida o dejar secuelas, por lo que debe diagnosticarse precozmente y tratarse de forma adecuada. Casos clínicos: Presentamos dos recién nacidos que fueron diagnosticados tardíamente de hipertiroidismo neonatal, en la tercera semana de vida. En ambos casos la clínica estaba presente desde el nacimiento pero el antecedente materno había pasado desapercibido. Se trataba de mujeres con enfermedad de Graves tratadas con ablación tiroidea con radioyodo años atrás, constando únicamente en sus historias clínicas que eran hipotiroideas en tratamiento sustitutivo con levotiroxina. Los dos niños habían sido prematuros y presentaban desde el nacimiento irritabilidad, insomnio y escasa ganancia de peso. El caso número 1 incluso fue hospitalizado desde el 5º día de vida hasta el día 16º por hipertermia y deshidratación hipernatrémica, constatándose una taquicardia sinusal persistente sin filiar tras estudio cardiológico. El diagnóstico de hipertiroidismo no llegó hasta el día 18º de vida al conocerse los resultados del cribado universal de hipotiroidismo (TSH suprimida). El caso 2 fue diagnosticado por un familiar médico. La Tabla presenta más datos de los casos. Comentarios: Las mujeres embarazadas con enfermedad de Graves en actividad, así como las tratadas previamente mediante tiroidectomía o radioyodo, deben someterse al menos a una determinación de Ac RTSH entre las semanas 22ª y 26ª de gestación y si estos niveles están por encima del triple de los normales, ser advertidas del riesgo de hipertiroidismo neonatal transitorio y adecuadamente dirigidas. En circunstancias ideales, otra determinación anterior, en el primer trimestre del embarazo, sería recomendable para vigilar la posible afectación fetal. El obstetra, el pediatra y la matrona también deben conocer esta enfermedad y diagnosticarla precozmente en el hijo para evitar sus potenciales secuelas. En los casos en los que no se disponga de determinación de Ac RTSH maternos se debe actuar como si estos fueran elevados.
P2/d3-128
HIPOTIROIDISMO CONGÉNITO ASOCIADO A CARDIOPATÍA CONGÉNITA A. Herranz Cecilia(1), J.M. Lezana Rosales(1), B. Cano Gutierrez(2), J.C. Moreno Navarro(1) (1) Instituto de Genética Médica y Molecular (INGEMM). Hospital Universitario la Paz, Madrid; (2) Hospital Ramón y Cajal. Madrid Introducción: El hipotiroidismo congénito (HC) se ha asociado a la presencia de otros defectos congénitos, entre los que destacan las anomalías cardíacas. Esta relación puede estar debida a la proximidad en el desarrollo embrionario del corazón y el tiroides y a que ambas estructuras expresan genes comunes, como NKX2-5, TBX5 o TBX1. Objetivos: Identificar la presencia de cardiopatía congénita en una cohorte de pacientes con HC, subclasificar en grupos en base a características fenotípicas y analizar su base molecular. Pacientes y Métodos: Caracterización del fenotipo cardiológico y tiroideo en una cohorte de 50 pacientes con HC. Búsqueda de mutaciones en los genes de NKX2-5, TBX5 o TBX1 por amplificación y secuenciación. Resultados: Hasta el momento se han podido fenotipar completamente alteraciones tiroideas y cardiológicas en 16 pacientes. El HC en el grupo estudiado era debido a disgenesia tiroidea en un 43,3% de los casos (hipoplasia tiroidea: 37% y hemiagenesia: 6,3%). El 50,4% de los pacientes presentaron dishormonogénesis y un 6,3% HC central. Entre las alteraciones cardiológicas destacan los defectos septales, en un 68,75% (comunicación interventricular y/o interauricular). Los defectos truncales y de la aorta correspondieron al 25% de los casos, obstrucción del tracto de salida: 12,5%, bloqueo auriculo-ventricular: 12,5%, miocardiopatía: 6,3% e hipoplasia del ventrículo izquierdo: 6,3%. Ningún paciente mostró anomalías derecha-izquierda. De los 16 casos, 6 se pudieron encuadrar dentro de un síndrome concreto (S.Williams, S.Holt-Oram, S.Down y CATCH22), los 10 restantes no se encuadraron en ningún síndrome conocido. Uno de los pacientes con HC bociogéno por dishomonogénesis muestra la mutación heterozigota IVS4-2A Conclusión: El tipo de cardiopatía más frecuente asociada a HC fueron los defectos septales, concretamente un 52% de comunicación interventricular. Es esencial realizar un correcto y completo estudio fenotípico en pacientes con HC, ya que esto permite orientar el estudio genético dirigiéndolo a genes candidatos a mostrar alteraciones (NKX2.5, GATA4, TBX5, TBX1).
P2/d3-129
DIAGNÓSTICO DIFERENCIAL DE MASA A NIVEL DE LA LENGUA J.J. Momblán De Cabo, Jl. Gomez Llorente, A. Bonillo Perales Hospital Torrecardenas, Almeria Introducción: Una masa a nivel de la base de la lengua puede estar motivada por una multitud de causas como son hemangiomas, adenoides, amigdala lingual, lipomas , carcinomas y el tiroides lingual. El tiroides lingual es un alteración poco común, de origen embrionario, generada por una falta del descenso normal de la glándula tiroides. Su incidencia es de una de cada 100.000 personas y es más frecuente en mujeres. Caso clínico: Presentamos el caso de un niño de 5 años que es derivado desde urgencias a la que acudió por molestias en la garganta, sin otra sintomatología. En la exploración destaca una masa en la base de la lengua de unos 3x3 cm2 roja que ocluye parcialmente la arcada faringea. Se realizó analitica basal con perfil tiroideo siendo estos resultados normales y se solicitó una Gammagrafia que confirmó la existencia de dicho tiroides. Debido al tamaño de la masa se inició tratamiento con hormona tiroidea consiguiendo una reducción significativa de dicha masa tiroidea. Discusión: Este transtorno raro ocurre más en mujeres con una proporción de 1:4, el 70% de los casos cursa con diversos grados de hipotiroidismo, que no presenta nuestro caso y hasta en un 10% se encuentra de forma casual en personas sanas. El diagnóstico diferencial incluye hemangiomas, adenoides, amigdala lingual, lipomas y carcinomas. Los examenes complementarios como TAC, ecografía y la gammagrafía hacen un diagnóstico relativamente fácil en la mayoría de los casos. El tratamiento debe individualizarse en casos asintomáticos se puede realizar tratamiento conservador expectante con estudio de función tiroidea periódica. En caso de tamaño moderado (como es nuestro caso), se ha descrito tratamiento con hormona tiroidea para reducir dicho tamaño y en caso de originar alguna complicación (obstrucción vía aérea, sangrado... etc) tratamiento quirúrgico si no ha respondido al médico.
P2/d3-130
EVOLUCIÓN NATURAL DE LA HIPERTIROTROPINEMIA AISLADA EN ESTUDIO RETROSPECTIVO DE 2 AÑOS DE DURACIÓN G. Roca Gardeñas, J. Pérez Sánchez, M. González Moreno, J. Rivera Luján, R. Corripio Collado Unidad de Endocrinología Pediátrica. Servicio de Pediatría. Hospital de Sabadell. Corporació Sanitària Parc Taulí. Institut Universitari Parc Taulí-UAB, Sabadell/Barcelona Introducción: La hipertirotropinemia se caracteriza por elevación de la hormona estimulante del tiroides (TSH) con niveles normales de hormonas tiroideas (T4l). Se desconoce con exactitud el riesgo de desarrollar hipotiroidismo en estos pacientes. Objetivo: Describir la evolución natural de los pacientes con hipertirotropinemia diagnosticados y controlados en nuestro centro durante 2 años. Material y métodos: Identificación de los pacientes derivados entre 2010 y 2011 por hipertirotropinemia con anticuerpos tiroideos (AT) negativos. Obtención de valores séricos de TSH, T4l y AT al diagnóstico y durante su seguimiento. Según evolución analítica, clasificación de los pacientes en: hipertirotropinemia transitoria (TSH<5 mUI/l), hipertirotropinemia persistente (TSH>5 mUI/l) o desarrollo de tiroiditis (AT positivos). Análisis de resultados de ecografía tiroidea y necesidad de tratamiento. Resultados: Se reclutó una muestra de 46 pacientes (27 niños) con una edad media 7,65±4,13 DE años (rango 10 meses-15,5 años). El valor medio inicial de TSH y T4l fueron 7,29±1,88 DE mUI/l y 1,20±0,15 DE nmol/l respectivamente. Ningún paciente presentaba AT positivos al diagnóstico. 28/46 pacientes (60,9%) presentaron normalización de los niveles de TSH en una media de 9,8±6,4 meses. Ésta se evidenció en el 36% de casos en los primeros 6 meses de seguimiento, y en el 83,3% en los primeros 12 meses. 16/46 pacientes (34,8%) persistieron con elevación de TSH durante un seguimiento de entre 8 y 43 meses. Dos de ellos recibieron tratamiento substitutivo por TSH >10 mUI/l. Por último 2/46 (4,3%) desarrollaron una tiroiditis con función tiroidea normal. En 15 pacientes se realizó ecografía tiroidea con resultado normal. Los valores iniciales de TSH en los pacientes con normalización final fue 6,54±1,4 DE y en los que persiste elevación 7,89±1,67 DE mUI/l, sin observarse diferencias estadísticamente significativas, así como tampoco se observan en edad o IMC entre los dos grupos. Conclusión: Más de la mitad de los pacientes de la muestra de estudio presentaron una normalización de los niveles de TSH. La normalización se produjo en los primeros 12 meses en casi todos ellos. Los valores iniciales de TSH no parecen ser un factor predictivo de la evolución posterior.
P2/d3-131
LEUCOPENIA, UN EFECTO ADVERSO POCO COMUN EN EL TRATAMIENTO CON ANTITIROIDEOS EN LA ENFERMEDAD DE GRAVES, A PROPÓSITO DE UN CASO A.D. Alcalde de Alvare, M. Magdalena Hawkins Solis, J. Yebra Yebra, M. Pacheco Cumani, A. Cañete Diaz Hospital Infanta Sofia, San Sebastián de los Reyes Madrid Introducción: La Enfermedad de Graves es una entidad poco frecuente en la infancia. Su incidencia no está bien descrita, la prevalencia es de 0,02% con una relación mujer/varón 6:1 y suele ser la responsable del 90-95% de los casos de hipertiroidismo en la infancia. El tratamiento inicial se basa fundamentalmente en antitiroideos, pero el índice de remisiones de estos fármacos en general es bajo y no están exentos de efectos adversos, así que es frecuente que los pacientes precisen evolutivamente tratamiento ablativo con cirugía o radioyodo. Materiales y métodos: Presentamos el caso de una niña de 10 años que consulta por un cuadro de hipertiroidismo (TSH <0.004 UI/L, T4l 3.49 ng/dl y T3l 13,17ng/dl), detectado en analítica solicitada por su pediatra de atención primaria, por pérdida de peso, nerviosismo e insomnio. Se inició tratamiento antitiroideo con tiamazol, precisa dosis máximas de 0.31 mg/kg/día y se logra eutiroidismo que asocia mejoría de los síntomas asociados a las 10 semanas de tratamiento. Evolutivamente se confirman anticuerpos antitiroideos, y antitiroglobulina y TSI positivos, con ecografía tiroidea compatible con tiroiditis. La evolución clínica y analítica permiten ir bajando progresivamente la dosis de antitiroideo hasta 0,1 mg/kg/día al séptimo mes de tratamiento, momento en el que se evidencia leucopenia (4.000 leucocitos/microL) y leucopenia (1.200 neutrófilos/microL). Se suspende el tratamiento y se repite control analítico tres semanas más tarde comprobándose la resolución de la citopenia, pero con reaparición del hipertiroidismo tanto clínico cómo analítico. Resultados: después de evaluar la posible evolución de los efectos adversos del tiamazol, frente a otros antitiroideos o un tratamiento ablativo se decide una actitud expectante reiniciándose el tratamiento con tiamazol a la dosis previa con controles clínicos y analíticos seriados. Conclusiones: El escaso número de casos registrados en la literatura de los efectos secundarios de los antitiroideos en niños, obliga a que las decisiones terapéuticas en la enfermedad de Graves deban basarse, con frecuencia, en la valoración clínica junto con estudios realizados en población adulta.
P2/d3-132
HIPERTIROIDISMO CONGÉNITO POR ENFERMEDAD DE GRAVES-BASEDOW (EGB). EXPERIENCIA CON MONOTERAPIA CON LUGOL G. Membrillo Lucena(2), J. Cayrol Cancela(1), N. Navarro Patiño(3), B. Hernández Rupérez(3), M.D. Rodríguez Arnao(1), M.D. Rodríguez Sánchez(1) (1) Unidad de Metabolismo y Endocrinología Infantil. Hospital General Universitario Gregorio Marañón, Madrid; (2) Hospital Materno-Infantil de Badajoz. CHU Badajoz; (3) Hospital General Universitario Gregorio Marañón. Madrid Introducción: El hipertiroidismo congénito es una entidad infrecuente en Pediatría (1/50.000 niños). La causa habitual es el paso de autoanticuerpos trasplacentarios de una madre con enfermedad de Graves- Basedow con gran actividad estimulatoria (1-2% de gestantes con EGB), y suele corresponder a una situación transitoria en el niño. Casos clínicos: Caso 1: RN de 31+5 semanas de edad gestacional (EG). Antecedentes prenatales: madre diagnosticada de EGB, tiroidectomizada y levotiroxina sustitutiva posterior. Ecografías prenatales:CIR, hipertensión pulmonar y taquicardia fetal. Exploración: PRN:1660 gr (P50), LRN:43 cm (P75), aspecto desnutrido, exantema macular violáceo, patrón colestásico (ictericia, hipertransaminasemia e hiperbilirrubinemia conjugada), taquicardia sinusal, bocio, hiperexcitabilidad y aumento del apetito. Pruebas complementarias: TSH: 061 mU/L (N:05-45), T4libre: 46 ng/dL (N:06-14), anticuerpos antitiroideos: negativos,ecografía cervical: aumento tiroideo. Sos-pechándose infección congénita por citomegalovirus se inicia tratamiento con ganciclovir (suspendido tras serología negativa) y se diagnosticó de hipertiroidismo congénito por EGB materna, iniciándose tratamiento con lugol por contraindicar la hepatopatía el uso de tionamidas. Actualmente asintomática y con normofunción tiroidea. Caso 2: RN de 34+4 semanas de EG. Antecedentes prenatales: madre diagnosticada de EGB, tratada con I131 y actualmente metimazol y levotiroxina. Anticuerpos estimulantes para receptor TSH positivos. Ecografías prenatales: taquicardia y bocio fetal. Exploración: PRN:2320 gr (P50), LRN: 49 cm (P97). Al 8º día de vida:taquicardia sinusal, irritabilidad, deposiciones semilíquidas, febrícula y bocio. Pruebas complementarias: TSH: 04mUI/dl (N:5-45), T4 libre:56 ng/dL (N:6-14), anticuerpos antitiroideos:positivos. Ecografía cervical: aumento tiroideo. Se diagnostica de Hipertiroidismo Congénito por EGB materna e inicia tratamiento con lugol. Continúa con el mismo, permaneciendo asintomático y controles hormonales normales. Conclusiones: • El hipertiroidismo congénito es un desorden infrecuente con importantes repercusiones influyentes en el desarrollo postnatal (alteraciones hemodinámicas y del crecimiento). • La presentación clínica puede ser similar a la de infecciones virales connatales. • Supone una urgencia, requiriendo un diagnóstico y tratamiento precoz, siendo imperativo una sospecha clínica e identificación prenatal de factores de riesgo. • El tratamiento precoz suele remitir la clínica en pocas semanas o meses. • La monoterapia con lugol, poco descrita hasta ahora, resultó eficaz en ambos casos, sin presentar fenómeno de escape, planteando una nueva estrategia terapéutica.
P2/d3-133
SÍNDROME DE RESISTENCIA A HORMONAS TIROIDEAS: A PROPÓSITO DE UN CASO D. Calvo Martínez, J.M. Martos Tello, A. Escribano Muñoz, A. Gutiérrez Macías Endocrinología Infantil-Hospital Universitario Virgen de la Arrixaca, El Palmar (Murcia) Introducción: La resistencia a hormonas tiroideas es un grupo de síndromes de causa genética caracterizados por la disminución de la sensibilidad tisular a estas hormonas por mutaciones en el gen THRB. Caso clínico: Lactante de 7 meses remitida a nuestra consulta por elevación de T4 libre con ausencia de supresión de TSH: TSH 1,55µUI/ml; T4 libre 37pmol/l. Antecedentes familiares: abuela paterna, padre y tío paterno con alteración tiroidea similar, todos ellos asintomáticos y sin tratamiento. Antecedentes personales: embarazo, parto y período neonatal sin incidencias. Acude a Estimulación temprana por retraso psicomotor: sonrisa social a los 5 meses; no sostén cefálico ni sedestación a los 7 meses. Resto no relevante. Exploración física: Peso en p7, longitud en p26, PC en p98. No bocio. Hipotonía axial. No sostén cefálico, no sedestación. Exploraciones complementarias: se solicita estudio genético de gen THRB, con alteración genética c.728G>A (p.Arg243Gln) en heterozigosis en el exón 7, confirmando el diagnóstico clínico de resistencia a hormonas tiroideas. Ecografía tiroidea normal. Evolución: se inicia tratamiento con levotiroxina, con importante mejoría clínica, comprobando al mes del inicio adquisición de sostén cefálico y sedestación estables junto a mejoría del tono muscular. Discusión: La resistencia a hormonas tiroideas es una enfermedad genética autosómica dominante que se ha de sospechar ante concentraciones altas de hormonas tiroideas en suero junto a TSH normal o elevada. Una misma mutación puede dar lugar a distintas formas clínicas entre familias o incluso en pacientes pertenecientes a la misma familia. Aunque la mayoría de pacientes no requieren tratamiento, en los casos con signos de hipotiroidismo tisular (como retraso de desarrollo en niños) es necesario el tratamiento con levotiroxina incluso a dosis altas. |
| References |