
| Rev Esp Endocrinol Pediatr 2013;4 Suppl(1):142-143 | Doi. 10.3266/RevEspEndocrinolPediatr.pre2013.Apr.180 |
| Genética |
| Sent for review: 11 Apr. 2013 | Accepted: 11 Apr. 2013 | Published: 2 May. 2013 |
| Congreso SEEP |
| Correspondence:Congreso SEEP E-mail: seep@seep.es |
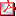 Figura - O3/d3-024 Figura - O3/d3-024 |
|
|
O3/d3-024 HETEROGENEIDAD CLÍNICA EN FUNCIÓN DE ALTERACIÓN GENÉTICA EN HIPERINSULINISMO. C. Fernández Ramos1, A. Vela Desojo2, R. Martinez Salazar2, M. Ortiz Espejo2, L. Castaño González2, Grupo Español de Hiperinsulinismo Congénito. (1) Endocrinología Infantil. Hospital Universitario Basurto, Bilbao/Vizcaya. (2) Grupo Investigación Endocrinología y Diabetes. Hospital Universitario Cruces, Baracaldo. Introducción: La secreción de insulina por la célula beta pancreática de una manera persistente ocasionando hipoglucemias severas se conoce como hiperinsulinismo congénito (HC). Existe gran heterogeneidad clínica y genética en esta entidad. Se conocen mutaciones en 8 genes distintos. Objetivos: Caracterizar clínica y genéticamente 49 familias con HC e intentar establecer una relación genotipo-fenotipo. Materiales y métodos: Se han estudiado en 52 pacientes con HC procedentes de 49 familias independientes, los genes: ABCC8, KCNJ11, GCK, HNF4A, GLUD1 por PCR y secuenciación automática directa. Los datos clínicos se recogieron en un cuestionario. Resultados: Se han identificado mutaciones en el 52% (27/52) de los pacientes. De ellos el 36% presentan mutaciones inactivantes en los canales de KATP: 18 en ABCC8 y 1 en KCNJ11. El resto presenta mutaciones en heterocigosis en otros genes: 4 casos mutaciones activantes en GCK, 3 casos con hiperamonemia y comienzo en edad lactante mutaciones en GLUD1 y 1 caso con macrosomía y madre diabética una mutación en HNF4A. Un 88% del total recibieron diazóxido con diferente respuesta (gráfica 1). Se realizó pancreatectomía en 11 casos (8 con mutación en ABCC8 y 3 sin genética). En aquellos con inicio precoz de la hipoglucemia (29), el 53% tienen mutación en los canales de KATP, 39% genética negativa, un 38% fue asintomática mientras que la hipotonía fue la manifestación clínica más frecuente. 3 fallecidos y el 24 % presentan secuelas. La convulsión es la clínica más frecuente de los que inician siendo lactantes, las mutaciones en GLUD se encuentran en este grupo etario. El inicio del HC con más de un año de edad se presentó en 4 casos, tres con alteración en GCK, todos con clínica leve y evolución sin secuelas. El % de casos con peso elevado al nacimiento es mayor si la genética es conocida (40% frente 16%). Conclusiones: Las mutaciones en ABCC8 son la causa más frecuente de HC. El inicio del cuadro es más tardío y leve en los casos con alteraciones en GCK y GLUD1 frente a los de ABCC8 y KCNJ11. La respuesta al diazóxido varía dependiendo del defecto genético. |
| References |
 |
 |
 |
 |
 |
 Print
Print View / Download PDF
View / Download PDF  English version
English version Comments
Comments Add Favorites
Add Favorites